Электрохимические
методы анализа
В химическом анализе применяют около десятка разных электрохимических
методов. Они имеют много общего. Во всех случаях анализируемую пробу переводят
в раствор и опускают в него два электрода. Система раствор - электроды представляет
собой электрохимическую ячейку. После установления равновесия измеряют с
помощью подходящего прибора аналитический сигнал, в данном случае -
характеристику ячейки, зависящую от
содержания определяемого компонента в исследуемом растворе.
Электрохимические методы классифицируют по природе аналитического сигнала.
Так, в ходе анализа можно измерять потенциал одного из электродов (потенциометрия), сопротивление ячейки
или электропроводность раствора (кондуктометрия).
Во многих случаях на электроды накладывают внешнее напряжение, после чего
измеряют силу тока, проходящего через раствор (вольтамперометрические методы, в частности полярография). При этом на поверхности электродов протекают
окислительно-восстановительные реакции, то есть идет электролиз раствора. Если провести электролиз
до конца и измерить количество электричества, пошедшего на окисление (или на
восстановление) определяемого вещества, можно рассчитать массу этого вещества.
Такой метод называют кулонометрией.
Иногда содержание определяемого вещества рассчитывают по привесу электрода, т.
е. по массе выделившегося на нем продукта электролиза (электрогравиметрия).
Электрохимические методы анализа были созданы в конце XIX – начале XX века на основе
достижений физики и физической химии. Примером могут быть закономерности
процесса электролиза, установленные Майклом Фарадеем в середине XIX века и ставшие затем основой
методов кулонометрии и электрогравиметрии. Теоретические аспекты и
аналитические возможности электрохимических методов были глубоко исследованы в
XX веке.
Электрохимические методы довольно селективны (кроме кондуктометрии),
поэтому с их помощью количественно определяют одни элементы в присутствии
других, раздельно определяют разные
формы одного элемента, делят сложные
смеси и идентифицируют их компоненты, а также концентрируют некоторые
микропримеси. Электрохимические методы широко применяют для контроля состава
природных и сточных вод, почв и пищевых продуктов, технологических растворов и
биологических жидкостей. Соответствующие методики не требуют сложного
оборудования, в них не используются высокие температуры и давления. Разные
электрохимические методы различаются по чувствительности, точности,
экспрессности и другим показателям, а потому хорошо дополняют друг друга.
Потенциометрический анализ
Принцип метода.
Аналитическим сигналом в потенциометрии является электродный потенциал.
Зависимость потенциала электрода от состава раствора была установлена на основании термодинамических функций немецким
физикохимиком Вальтером Нернстом (в будущем – лауреатом Нобелевской премии) в
![]() (1),
(1),
Если в растворе находится лишь окисленная форма вещества (восстановленную
представляет металлический электрод), концентрация окисленной формы - С моль/дм3, формулу (1) можно
упростить:
![]() (2).
(2).
Электроды и измерение их
потенциалов. Электрод, у которого равновесный потенциал (Еинд) зависит от концентрации
определяемого вещества (иона), называют индикаторным.
Этот потенциал
должен хорошо воспроизводиться при повторных измерениях, быстро устанавливаться
и, что наиболее важно, не должен зависеть от концентрации других компонентов
раствора. Второй электрод называют электродом
сравнения. Он должен
иметь постоянный потенциал Еср, не зависящий от состава
раствора. Значения Еср для обычно применяемых электродов сравнения
(хлорсеребряного, каломельного и др.)
точно известны и приведены в справочниках.
Потенциал электрода можно измерять только относительно какого-то другого
электрода. Именно поэтому в ходе анализа в раствор одновременно опускают два
разных электрода и, в качестве
первичного аналитического сигнала, измеряют разность потенциалов между
ними (ΔЕ). Измерения ведут с помощью специальных электронных
приборов – потенциометров.
Рассчитывать
точное значение Еинд в
ходе проведения анализа не обязательно, поскольку не только вторичный сигнал Еинд, но и непосредственно
измеряемая величина ΔЕ линейно связана с логарифмом
концентрации определяемого иона (аналита). Концентрацию аналита можно
определить, пользуясь градуировочным графиком, заранее построенным в координатах
ΔЕ
- lgC. Однако следует помнить, что подобные графики
линейны лишь в неизменных условиях измерения и лишь в определенном интервале
концентраций аналита.
Мембранные (ионселективные) электроды. В индикаторных электродах мембранного типа, применяемых в анализе, мембрана
отделяет исследуемый раствор от вспомогательного раствора внутри электрода. На
обеих поверхностях такой мембраны идут процессы ионного обмена между материалом
мембраны и растворами, устанавливаются ионообменные равновесия и соответствующие
им потенциалы. Если составы «внутреннего» и «внешнего» растворов различны,
возникает разность этих потенциалов, тем большая, чем сильнее отличается по
составу исследуемый раствор от внутреннего (состав внутреннего раствора
известен и строго постоянен). Материал мембраны и способ ее предварительной
обработки подбирают так, чтобы на электродный потенциал влияли ионы какого-то
определенного вида (например, Н+), а все остальные ионы влияли бы
как можно меньше. Поэтому мембранные электроды получили еще одно название – ионселективные электроды (ИСЭ). Когда
ИСЭ опускают в анализируемый раствор, создается электродный потенциал,
зависящий от концентрации только определяемых ионов
Первым электродом мембранного типа
стал созданный в
|
|
|
|
Рис.1. Схема стеклянного электрода 1 -
серебряная проволока 2 - 3 -
стеклянная мембрана |
Рис.2. Зависимость
потенциала стеклянного электрода от рН раствора (при 250С) |
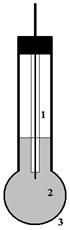
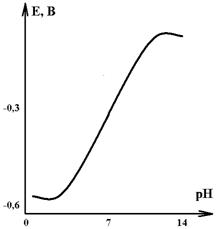
Рассмотрим схематическое устройство стеклянного
электрода (рис.1). Роль мембраны в данном случае выполняет тонкая стенка
стеклянного шарика, внутри которого находится
Меняя состав стекла и структуру мембраны, можно
получить стеклянные электроды, селективные относительно ионов Na+ или других щелочных металлов. Для селективного
определения других металлов (например,
ионов Cu2+, Ag+,
Cd2+, Hg2+) применяют кристаллические мембраны, например, халькогенидные, то есть
содержащие сульфиды металлов. Многие анионы определяют с помощью жидкостных
мембран. В этом случае ионообменный материал (вязкая органическая жидкость)
распределен внутри эластичной полимерной пленки, отделяющей внутренний раствор
от внешнего. Кроме мембранных электродов, к числу ИСЭ относят и некоторые
другие электроды – газочувствительные,
ферментные, полевые транзисторы и т.п.
Потенциометрический анализ включает два основных
варианта - прямую потенциометрию (ионометрию)
и потенциометрическое титрование.
Прямая потенциометрия – это определение содержания аналита непосредственно по величине аналитического
сигнала, т.е. потенциала индикаторного электрода. Метод прямой потенциометрии – экспрессный и легко автоматизируемый,
он не требует дорогой и сложной аппаратуры. Поэтому метод широко применяют в
практике, в частности, при исследовании состава природных и сточных вод, почв,
технологических растворов; в анализе пищевых продуктов, биологических жидкостей
и т.п. Прямую потенциометрию используют для определения растворенного в воде кислорода,
фторидов и цианидов в сточных водах, нитратов в почвах и пищевых
продуктах, а также для определения
некоторых органических веществ. Важнейшим применением потенциометрии является
измерение рН с использованием стеклянного электрода. Величина рН – важный
показатель качества воды, пищевых
продуктов, лекарственных и косметических препаратов и других товаров.
Непрерывно контролировать величину рН приходится при проведении многих
технологических процессов. В этом случае потенциометрические сенсоры помещают
внутри соответствующих реакторов, в трубопроводах и т.п.
Метод прямой потенциометрии имеет и ряд ограничений. Основное -
сравнительно невысо-кая точность. Как и в других электрохимических методах,
твердые пробы приходится переводить в раствор, что существенно удлиняет анализ
и приводит к дополнительным погрешностям. Потенциометрическим методом часто не
удается определять содержание микропримесей. Еще одно ограничение -
необходимость иметь в лаборатории множество индикаторных электродов, ведь для
каждого определяемого вещества нужен свой электрод. В настоящее время аналитическое
приборостроение выпускает около тысячи разных ИСЭ для потенциометрического
анализа (различных типов, не только мембранных), но для подавляющего
большинства веществ подходящие индикаторные электроды пока еще не созданы.
Потенциометрическое
титрование. В этом случае погружают в исследуемый раствор электроды и
постепенно добавляют к этому раствору какой-либо титрант. Из-за протекающей в
ходе титрования химической реакции потенциал индикаторного электрода постоянно
меняется. Проводя потенциометрическое титрование, измеряют величину ΔЕ
после добавления каждой порции титранта, а затем строят кривые титрования.
Резкий скачок измеряемой величины должен наблюдаться вблизи точки эквивалентности.
Потенциометрическое титрование возможно даже в очень разбавленных растворах. Таким методом можно анализировать
смеси сложного состава, раздельно определяя в них содержание каждого
компонента. Например, в одной пробе можно раздельно определить содержание
хлоридов, бромидов и иодидов (на кривой аргентометрического титрования
смеси отчетливо видны три скачка). Можно
титровать мутные и окрашенные растворы.
В обоих
вариантах потенциометрического анализа применяют одни и те же электроды, те же
схемы измерений. Однако для потенциометрического титрования не требуется такая
высокая точность измерения потенциалов, как для прямой потенциометрии. Ведь
положение точки эквивалентности устанавливают не по абсолютным значениям
потенциала электрода, а по его изменениям в ходе реакции. Таким образом, для
потенциометрического титрования можно использовать упрощенные приборы и, тем не
менее, получать более точные результаты анализа. Не требуются в этом случае
эталонные растворы определяемых веществ. Важным преимуществом потенциометрического
титрования по сравнению с прямой потенциометрией (ионометрией) является и более
широкий круг определяемых веществ, так можно определять и те вещества, для которых еще не созданы подходящие
индикаторные электроды.
Потенциометрический
контроль может быть использован в любом варианте титриметрического анализа.
Надо только правильно выбрать индикаторный электрод: его потенциал должен быть
линейно связан с логарифмом концентрации титруемого вещества или титранта. Для
кислотно-основного титрования обычно применяют стеклянный электрод, в
аргентометрии – серебряный, в
редоксметрических методах – индифферентные электроды (обычно
платиновый), в комплексонометрии – различные ИСЭ. Электродом сравнения, как и в прямой
потенциометрии, служит электрод 2-го рода – хлорсеребряный или каломельный. На
практике потенциометрическое титрование широко применяют при определении серы и ее соединений в нефтепродуктах и
горных породах, а также в анализе технологических растворов, объектов окружающей
среды, лекарственных препаратов и биообъектов.
Конечно, потенциометрическое титрование – более длительная и трудоемкая
процедура, чем прямая потенциометрия. Однако титрование можно автоматизировать,
для этого вводят титрант с постоянной
скоростью, непрерывно измеряют ΔЕ
и записывают кривую титрования с помощью
самописца или компьютера.
Вольтамперометрические методы
Электрохимические методы, использующие зависимость силы тока, протекающего через
раствор, от величины приложенного к электродам напряжения, называют вольтамперометрическими. Исторически
первым таким методом стала полярография. Так
называют вольтамперометрический метод, в котором рабочим является ртутный
капающий электрод (РКЭ). Полярография была
создана в
Классическая
полярография. Аппаратура и принцип метода. Ртутный капающий
электрод (РКЭ) представляет собой капилляр, соединенный с резервуаром,
наполненным ртутью (рис. 3). Под действием силы тяжести ртуть по каплям
вытекает из капилляра, следовательно, рабочая поверхность электрода постоянно
обновляется. Второй электрод - ртуть, находящаяся на дне электрохимической
ячейки. На электроды от внешнего
источника подают постоянное по знаку, но постепенно увеличивающееся по
величине напряжение. В полярографической ячейке РКЭ является катодом, а донная ртуть – анодом.
|
|
Рис.3 Схема полярографических измерений 1- ртутный капающий электрод 2- донная ртуть 3- капля ртути Г- гальванометр Б- внешний источник питания R-
переменное сопротивление |

Через ячейку с исследуемым раствором
идет постоянный ток. Зависимость силы тока от величины приложенного
напряжения называют вольтамперной кривой
или полярограммой. Если бы на поверхности электродов не шли
окислительно-восстановительные реакции, вольтамперная кривая представляла бы
собой (в соответствии с законом Ома) прямую линию. Но на реальных вольтамперных
кривых Гейровский обнаружил ступени («полярографические волны»). Положение и высота
каждой волны хорошо воспроизводились при повторении опытов и зависели от
состава раствора. Необходимо было
понять, почему появляются эти загадочные
волны, каким именно веществам они
соответствуют и как можно использовать обнаруженный эффект.
Вскоре Гейровский установил, что каждая из полярографических волн
соответствует восстановлению одного из
растворенных веществ-окислителей (в частности, компонентов анализируемой
пробы). Такие вещества электрохимики назвали деполяризаторами. Ими могли быть катионы металлов или водорода,
некоторые органические вещества, растворенный кислород, а также молекулы воды.
Если в растворе есть несколько деполяризаторов-окислителей с разными
потенциалами восстановления, то при постепенном увеличении напряжения, подаваемого
на ячейку, они будут восстанавливаться последовательно, и на кривой зависимости
силы тока от напряжения появятся не одна, а несколько волн.
Вольтамперные кривые. Рассмотрим (рис.3) вольтамперную кривую раствора,
содержащего один деполяризатор, например, ионы Cd 2+. Высота волны ,
отрезок ВС, зависит от концентрации
ионов в растворе, а величина потенциала полуволны – Е1/2 является
качественной характеристикой, которая зависит только от природы иона. Таким
образом, по величине Е1/2 можно провести качественный анализ, т.е., установить, какие ионы присутствуют в
растворе, а по высоте волны – узнать, какова их концентрация. Для нахождения
концентрации иона в исследуемом растворе предварительно строят градуировочный
график по ряду стандартных растворов с точно известными концентрациями ( рис.4)
Id
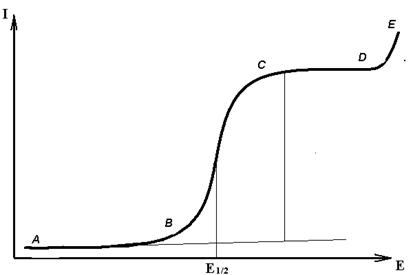
Рис.3 Полярограмма
раствора Cd(NO3)2
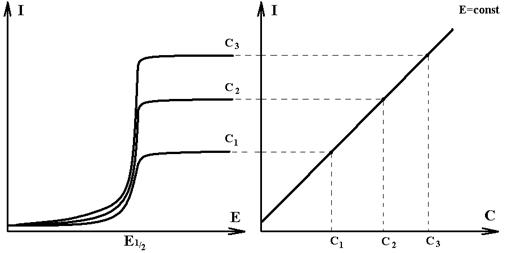
А Б
Рис.4. Полярограммы растворов Cd(NO3)2 разной
концентрации (А)
и градуировочный график
для определения ионов Сd 2+ (Б).
Для стандартных условий значения Е1/2 разных деполяризаторов приведены в справочной
литературе. Эти данные можно использовать в качественном анализе для
идентификации деполяризаторов. Значения Е1/2 также используют, чтобы оценить возможность
раздельного определения нескольких деполяризаторов. Например, в нейтральной
среде потенциалы полуволн цинка и никеля почти совпадают; значит,
раздельное определение этих ионов в их
смеси невозможно (на полярограмме смеси будет лишь одна волна, соответствующая
суммарной концентрации катионов никеля и цинка). А вот в среде аммиачного
буферного раствора потенциалы полуволн
тех же ионов различаются почти на 0,4 В, так как процесс
восстановления Zn2+ из-за комплексообразования смещается в более
отрицательную область. В этих условиях на вольтамперной кривой будут
наблюдаться две волны, раздельное
определение цинка и никеля окажется
вполне возможным.
Применение полярографии в анализе. Достоинства и недостатки полярографии как
аналитического метода. Еще в 1925 году Гейровский и Шиката
сконструировали самопишущий полярограф,
и этот прибор сразу же стал применяться в аналитических лабораториях, преимущественно
для определения малых
количеств меди, свинца,
кадмия, цинка и других металлов в сплавах и минералах. Длительные и трудоемкие
операции предварительного разделения компонентов теперь не требовались. Можно
было порознь определять различные формы
одного и того же элемента (например, Cr3+ и CrO42-).
Позднее полярографию стали использовать еще шире - для
определения неметаллов и органических веществ. Метод вызвал
всеобщий интерес, ведь полярограф был первым
автоматизированным аналитическим прибором, пригодным для многоэлементного анализа – как
качественного, так и количественного – самых разных объектов. Классическая
полярография давала очень высокую сходимость
результатов. Метод был
селективным, чувствительным (нижняя граница определяемых концентраций – 10-5
- 10-
Современные полярографы имеют гораздо большие возможности, чем полярограф
Гейровского. В частности, кроме обычных вольтамперных кривых, они позволяют
регистрировать производные этих кривых, например, в координатах ![]() . В этом случае на полярограмме смеси деполяризаторов
наблюдается ряд пиков. Положение каждого пика совпадает с потенциалом полуволны
на обычной полярограмме, а высота пика пропорциональна концентрации соответствующего
деполяризатора. При таком способе
регистрации повышается селективность анализа
– можно идентифицировать и раздельно определять компоненты, потенциалы полуволн
которых отличаются всего на 0,06 В. Что касается правильности и сходимости, то
эти параметры остаются примерно такими же, как и в классическом варианте полярографии.
. В этом случае на полярограмме смеси деполяризаторов
наблюдается ряд пиков. Положение каждого пика совпадает с потенциалом полуволны
на обычной полярограмме, а высота пика пропорциональна концентрации соответствующего
деполяризатора. При таком способе
регистрации повышается селективность анализа
– можно идентифицировать и раздельно определять компоненты, потенциалы полуволн
которых отличаются всего на 0,06 В. Что касается правильности и сходимости, то
эти параметры остаются примерно такими же, как и в классическом варианте полярографии.
В 50-х годах XX века практически одновременно
возникло несколько «неклассических» вольтамперометрических методов. От обычной
полярографии они отличались и по
виду рабочего электрода, и по способу измерения аналитического сигнала. К их
числу относят амперометрическое титрование, биамперометрию, дифференциальную и
осциллографическую полярографию, инверсионную вольтамперометрию,
переменнотоковую вольтамперометрию и некоторые другие методы. Наиболее широко в
исследовательских и контрольно-аналитических лабораториях применяют амперометрическое титрование и
инверсионную вольтамперометрию (ИВА).
Амперометрическое титрование. Этот метод, разработанный известным американским
аналитиком И.Кольтгофом, обеспечивает высокую точность анализа.
В анализируемый раствор помещают
два электрода и подают на них напряжение, вызывающее появление
предельного диффузионного тока. На рабочем электроде происходит восстановление (либо окисление) определяемого
компонента, но так как сила тока невелика, заметного снижения концентрации компонента
за счет электрохимического процесса не происходит. Затем в раствор начинают
вводить подходящий титрант, который реагирует с определяемым веществом.
Предельный диффузионный ток при этом снижается, вплоть до достижения т.экв.
После т.экв. ток, идущий через ячейку, уже не меняется (рис. 5 кривая А). По
перегибу линейной кривой
амперометрического титрования находят положение т.экв, а затем
рассчитывают результат анализа по обычным формулам титриметрического анализа. Создавать аналитический сигнал, меняющийся в
ходе титрования, может как определяемое
вещество, так и титрант, а также продукт взаимодействия титранта с
определяемым компонентом. В этих случаях
также будут наблюдаться линейные
кривые титрования, но их форма будет иной (рис.5, кривые В и С).
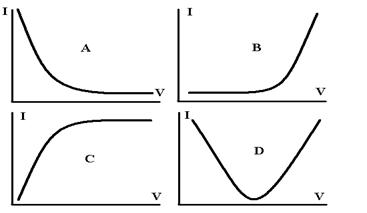
Рис.5. Кривые
амперометрического титрования разного типа
В некоторых случаях аналитический сигнал могут создавать сразу два
участника основной реакции. Например, на электроде могут окисляться и
определяемое вещество, и титрант. Тогда кривая титрования будет соответствовать
кривой D на
рис.5.
Амперометрическое титрование – более простой и универсальный метод, чем
классическая полярография. Таким способом можно определять даже те вещества,
которые не окисляются и не восстанавливаются на электродах. Метод весьма селективен. Высокая точность при измерении предельного
диффузионного тока не требуется,
важен лишь характер его изменения
в ходе титрования. Тем не менее амперометрическое титрование как метод
анализа дает высокую точность Но
чувствительность этого метода не лучше, чем
у классической полярографии.
Инверсионная
вольтамперометрия (ИВА) Этот метод детально разработан советскими
электрохимиками-аналитиками, в частности А.Г.Стромбергом и его учениками. Так как метод ИВА не требует
применения больших количеств ртути, он сравнительно безопасен. ИВА включает операцию предварительного
концентрирования определяемого элемента (как правило, металла). Поэтому нижняя
граница определяемых содержаний в этом методе составляет 10-9 - 10-
Чтобы провести анализ, пробу переводят в раствор, а затем при постоянном
перемешивании ведут электролиз этого раствора. В качестве катода при этом
используют неподвижную каплю ртути. В простейшем случае продолжительность
электролиза выбирают так, чтобы все катионы определяемого металла успели
восстановиться на катоде и накопиться там в виде амальгамы. Затем полярность
электродов меняют, капля ртути становится анодом, начинается быстрый процесс
анодного растворения накопившегося металла. Возникает ток растворения, который
и является аналитическим сигналом. Его величина пропорциональна концентрации
металла в капле. Так как объем капли во много раз меньше объема исходного
раствора, концентрация определяемого металла в капле во много раз больше
начальной. Во столько же раз усиливается аналитический сигнал. Кроме высокой чувствительности, метод ИВА
обладает и другими достоинствами. Можно одновременно накопить на катоде (в
капле ртути) сразу несколько металлов, а затем раздельно регистрировать токи их
анодного растворения (на полярограмме наблюдают несколько пиков при разных
потенциалах). По положению пиков можно опознать соответствующие металлы, а по высоте или площади пиков - рассчитать содержание каждого металла в
исходной пробе. Такой способ анализа обычно применяют для селективного определения следовых
количеств токсичных тяжелых металлов (свинец, кадмий и др.) в воде и пищевых продуктах, а также для контроля
микропримесей в химических реактивах и полупроводниковых материалах. Точность инверсионной
вольтамперометрии немного уступает
точности классической полярографии, но вполне достаточна для
решения многих химико-аналитических задач.