Спектроскопические методы
анализа
Классификация и основные принципы
спектроскопических методов
Все
спектроскопические методы основаны на взаимодействии атомов, молекул или ионов,
входящих в состав анализируемого вещества, с электромагнитным излучением. Это
взаимодействие проявляется в поглощении или испускании фотонов (квантов). В
зависимости от характера взаимодействия пробы с электромагнитным излучением
выделяют две группы методов - эмиссионные
и абсорбционные. В зависимости от того, какие
частицы формируют аналитиче- ский сигнал, различают методы атомной спектроскопии и методы молекулярной
спектроскопии.
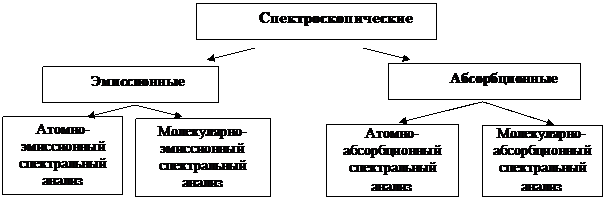
Рис. 1. Классификация спектроскопических методов
В эмиссионных методах анализируемая
проба в результате ее возбуждения излучает фотоны (кванты). Важнейшие
эмиссионные методы - атомно-эмиссионный
спектральный анализ (АЭС) и люминесцентный
анализ.
В
абсорбционных методах излучение постороннего источника пропускают через пробу, при этом
часть квантов избирательно поглощается атомами или молекулами. Важнейшие методы
этой группы - атомно-абсорбционный анализ
(ААС) и молекулярно-абсорбционная спектроскопия растворов. Последний метод
обычно называют спектрофотометрией
или фотометрическим анализом.
Абсорбционные методы, как и эмиссионные, используют и для обнаружения, и для
количественного определения веществ.
Кроме спектроскопических, известны и другие методы
анализа, основанные на оптических явлениях. В частности, в нефелометрии используют эффект рассеяния света, в рефрактометрии
- преломление светового потока; в поляриметрии
– вращение плоскости поляризации. Эти оптические методы к числу
спектроскопических не относят, поскольку они не связаны с поглощением или
излучением квантов.
Области электромагнитного спектра, применяемые в химическом
анализе. Электромагнитное
излучение может проявляться в разных формах: видимый свет, ультрафиолетовое
излучение, инфракрасное (тепловое) излучение, радиоволны, рентгеновские лучи,
гамма-лучи.
Если все фотоны (кванты) данного излучения
имеют одну и ту же энергию, излучение
называют монохроматическим, если их
энергии различны – полихроматическим.
Монохроматический свет имеет определенную длину волны. Полихроматическое
излучение характеризуется интервалом длин волн, в который входят все компоненты
данного излучения. Более полная характеристика полихроматического излучения – спектр. Он показывает распределение
интенсивности излучения по длинам волн
(или по энергиям, или по частотам).
Электромагнитное изучение охватывает очень
широкий интервал длин волн, а следовательно,
и энергий. Видимый свет соответствует лишь малой части этого интервала. В
химическом анализе применяют не все виды излучений. Широко используют
«оптический диапазон», границы которого - от 200 нм до 40 000 нм. В этот
диапазон входят три области:
·
ультрафиолетовая область (УФ) – 200-400 нм.
·
видимая область – 400-800 нм;
·
инфракрасная область (ИК) - от 800 до
40 000 нм.
Внутри каждой области иногда выделяют еще
более узкие участки, имеющие собственные названия. Так, в ИК-спектрах выделяют особую «область отпечатков пальцев».
Название связано с высокой информативностью этой области для опознания
индивидуальных органических веществ. В видимой области отдельные участки
характеризуют цветом излучения.
В XХ веке в анализе стали применять рентгеновские лучи и другие
виды излучений, не входящие в оптический диапазон. Для соответствующих методов анализа нужны
сложные и дорогие приборы,
которые пока что есть лишь в немногих лабораториях.
Спектры излучения и поглощения. Как правило, анализируемая
проба излучает и поглощает полихроматический свет, включающий кванты разной
энергии и разной длины волны. Однако для аналитика предпочтительнее измерять
испускание или поглощение света, в котором все кванты примерно одинаковы по
энергии, соответствуют одной длине волны. Чтобы выделить ее из
полихроматического излучения, нужно особое устройство – монохроматор. На рис.1. показана схема спектрального прибора с
призменным монохроматором.
Спектральные приборы, снабженные
монохроматорами, называют спектрометрами, спектрографами или стилоскопами, в
зависимости от используемого в них приемника излучения, то есть от того, какой
способ регистрации спектра (фотоэлектрический, фотографический или визуальный)
применяется в этих приборах. С помощью таких приборов можно зарегистрировать
спектр излучения или спектр поглощения исследуемой пробы.
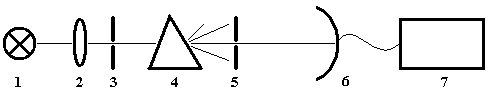
Рис.1. Схема
спектрометра с призменным монохроматором
1 – источник света, 2 –
фокусирующая оптика, 3 – входная щель, 4
– призма, 5 – выходная щель, 6 – приемник (фотоэлемент), 7 –
регистрирующее устройство (микроамперметр и т.п.).
Спектр
излучения пробы показывает, на каких длинах волн она преимущественно
излучает свет при возбуждении (например, при сильном нагревании). Спектр
излучения регистрируют в координатах:
интенсивность (I) излучения -
длина волны (l).
Спектр поглощения пробы показывает, на каких длинах волн она
преимущественно поглощает излучение внешнего источника. Такие спектры обычно
регистрируют в координатах A – λ,
где А - количественная характеристика
поглощения света на данной длине волны, называемая оптической плотностью.
Спектры поглощения и излучения
одного и того же вещества в некоторой области длин волн очень похожи. Чтобы
понять, почему это так, надо вспомнить, что атом может находиться только в
определенных состояниях, которым отвечают дискретные энергетические уровни. Переходя
под воздействием внешнего излучения в более возбужденное состояние, атом должен
приобрести дополнительную энергию DЕ за счет поглощения кванта. Поэтому в спектре поглощения
пробы на длине волны l,
соответствующей DЕ, появится линия (пик). В атомах данного элемента
возможны и другие энергетические переходы (с другими значениями DЕ). Все они реализуются одновременно, приводя к другим
линиям в спектре поглощения данного элемента.
Теперь рассмотрим атомы, которые уже переведены в
возбужденное состояние, например, под действием высокой температуры. Через
короткое время (10-7 – 10-8 сек) после возбуждения они
самопроизвольно возвращаются в основное состояние, излучая кванты. Энергии этих
квантов равны разностям энергий соответствующих состояний. Каждому переходу
соответствует некоторая длина волны, некоторая линия в спектре испускания.
Поскольку поглощение и испускание света
определяются одними и теми же энергетическими переходами, в спектрах поглощения
и излучения данного элемента наблюдаются одни и те же линии.
На рисунке 2
сопоставлены определенные участки спектров излучения и поглощения одного и того
же элемента, зарегистрированные в одинаковых условиях. Они очень похожи,
поскольку определяются однотипными энергетическими переходами в атомах данного
элемента.
А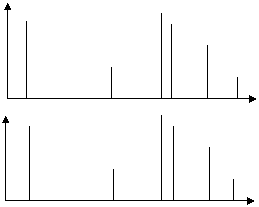
l, нм I l, нм
Рис.2. Атомные спектры поглощения (вверху) и излучения (внизу)
Особенности спектров разного типа и их аналитическое применение. Атомные спектры поглощения и
излучения, наблюдаемые во всем
оптическом диапазоне, определяются переходами электронов, относящихся к
наружным слоям («валентные электроны»). Таким образом, атомные спектры по своей
природе являются электронными, а
по внешнему виду - линейчатыми..
Положение спектральных линий в шкале длин волн и их относительную интенсивность
используют как идентификационные признаки в качественном элементном анализе.
Молекулярные спектры излучения или поглощения обычно не являются
линейчатыми. Вид молекулярных спектров в разных диапазонах длин волн различен,
поскольку различно происхождение соответствующих спектров. Спектры поглощения
молекул в видимой или УФ-области являются широкополосными.
Они дают сравнительно мало информации для
выяснения состава и структуры поглощающих молекул. Это мешает проведению
качественного анализа по спектрам в УФ- или видимой области.
Изучение молекулярных спектров –это важнейший способ
количественного химического анализа. Заметим, что количественное определение
какого-либо вещества по известной методике вовсе не требует регистрации полного спектра излучения (или
поглощения) пробы. Достаточно было бы измерить аналитический сигнал на заранее
выбранной длине волны. Спектры нужны для решения гораздо более сложных задач. А
именно:
Ø
По спектру индивидуального вещества выбирают ту длину
волны, на которой в дальнейшем, в ходе количественного анализа, будут измерять аналитический сигнал этого
вещества (I или А). Если для определения какого-либо элемента в
атомно-эмиссионном спектральном анализе используют наиболее интенсивные линии
эталонного спектра, то в молекулярно-абсорбционном (спектрофотометрическом)
анализе аналитический сигнал обычно измеряют на длине волны, соответствующей максимуму
на спектральной кривой.
Ø
Сопоставляя спектры предполагаемых компонентов пробы,
выясняют возможность определения одних веществ в присутствии других. Если
спектры компонентов пробы накладываются друг на друга, результаты анализа смеси
будут завышенными. Для снижения систематических
погрешностей, связанных с наложением спектров, созданы особые приемы измерений
и расчета результатов. Другие выходы из положения - маскирование или
предварительное отделение мешающих компонентов.
Атомно-эмиссионный
спектральный анализ
История
и принцип метода. Еще в древности было замечено, что цвет пламени меняется
при введении в него некоторых веществ. В XVIII веке этот эффект стали
использовать в анализе; в частности, по окраске пламени различали соду (Na2CO3)
и поташ (К2CO3). В XIX веке был установлен линейчатый
характер спектров пламени. Спектры начали фотографировать, определять длины
волн отдельных линий. Некоторые исследователи указывали, что, по наличию определенных
спектральных линий можно судить о присутствии в пробе тех или иных элементов.
Создателями спектрального анализа стали выдающиеся немецкие ученые – химик
Р.Бунзен и физик Г.Кирхгоф. В 50–70-е годы XIX века они вели совместные
исследования, используя спектроскопы собственной конструкции. В результате исследований
Бунзена и Кирхгофа был установлен качественный (элементный) состав многих
минералов и даже небесных тел; открыт ряд ранее не известных элементов (таллий,
индий и др.); созданы первые, еще не
очень точные способы количественного спектрального анализа.
На рубеже XIX и XX веков для
получения спектров стали применять электрическую дугу и искру, это позволило
определять и те элементы, которые не возбуждаются в пламени. Было доказано, что
спектральный анализ применим для обнаружения и определения элементов, независимо
от их степени окисления и от того, в составе каких химических соединений они
находились в исходной пробе. В 20-х годах XX века удалось значительно повысить
точность количественного анализа. Начался массовый выпуск спектральной аппаратуры.
Были созданы надежные методики атомно-эмиссионного спектрального анализа (АЭС)
для геологических и заводских лабораторий. В СССР АЭС стал основным способом
аналитического контроля в черной и цветной металлургии, а также в геологии.
Дальнейшее развитие метода связано с появлением новых источников возбуждения
(особенно индукционно связанной плазмы) и новых способов регистрации спектров,
а также с автоматизацией и компьютеризацией анализа.
Принцип метода довольно прост. Во
всех вариантах АЭС пробу вносят в источник возбуждения, где тем или иным
способом создается высокая температура (тысячи градусов). Образуется плазма
(совокупность возбужденных атомов, ионов и электронов). В ней последовательно
проходят следующие процессы:
·
испарение
пробы;
·
атомизация первоначальных продуктов испарения (молекул
или ионов);
·
возбуждение
образовавшихся атомов;
·
испускание
света возбужденными атомами.
Возникающее в ходе анализа полихроматическое
излучение пробы фокусируют и направляют на входную щель спектрального прибора
(рис.1), где оно разлагается в спектр и
регистрируется соответствующим приемником (фотоэлемент, диодная линейка,
фотопластинка и др.). Можно наблюдать спектр и визуально (именно так работали
Бунзен и Кирхгоф), однако это небезопасно для глаз. Для качественного анализа
полученный спектр сопоставляют с эталонными спектрами разных элементов. По
одному спектру пробы можно быстро и надежно обнаружить многие, а то и все
присутствующие в ней элементы. Для количественного анализа надо измерить
интенсивность излучения пробы на
некоторых заранее выбранных длинах волн.
На практике измеряют не саму интенсивность излучения (число квантов,
испускаемых пробой в единицу времени), а зависящие от нее другие величины
- фототок, абсолютное или относительное
почернение фотопластинки и др. По этим аналитическим сигналам и рассчитывают
содержания разных элементов, пользуясь
градуировочными графиками, заранее полученными
с помощью подходящих эталонов.
Приборы для спектрального анализа включают в себя три основных блока: блок
возбуждения, диспергирующее устройство и блок регистрации излучения. Разные
варианты АЭС различаются по способу возбуждения пробы и по способу регистрации
спектра.
Источники возбуждения. В качестве источников возбуждения применяют пламя,
электрическую дугу, искру, а также высокочастотную индуктивно-связанную плазму
(ИСП, ICP). Три первых источника являются «классическими». Недавно созданный
метод ICP только входит в практику работы аналитических лабораторий, но именно
он обеспечивает наилучшие результаты. В научных исследованиях используют также
импульсный разряд, микроволновой разряд, лазерное излучение и некоторые другие
источники плазмы.
Способы регистрации спектра. Фотографическая регистрация достаточно
проста по технике, доступна. Этот «классический» способ позволяет получать и измерять даже очень
слабые сигналы микропримесей. Одновременно регистрируются линии всех
компонентов пробы. Сфотографированные спектры можно долго хранить и в любое
время провести повторные измерения.
Схема спектрографа похожа на схему
спектрометра, показанную на рис. 1, но выходная щель и измерительное устройство
в данном случае не нужны. После разложения излучения пробы по длинам волн оно
направляется на фотопластинку, содержащую в своем поверхностном слое кристаллы
бромида серебра. В местах, куда попадет излучение, образуется металлическое
серебро. В результате проявления и закрепления фотопластинки ее почернение во
много раз усиливается. На пластинке остается
спектр пробы в виде ряда черных линий одинаковой высоты и ширины, но с
разной степенью почернения (картинка напоминает штрих-код товара). Все эти линии являются фотографиями входной
щели, сделанными на разных длинах волн, соответственно спектральному составу
излучения пробы. На одну и ту же
фотопластинку можно последовательно сфотографировать десятки спектров, размещая
их друг под другом.
Почернение аналитических линий на проявленной фотопластинке измеряют с
помощью вспомогательного прибора – микрофотометра.
Фотоэлектрическая регистрация основана на применении фотоэффекта. Как было установлено
на рубеже XIX и XX веков, фототок приблизительно пропорционален интенсивности
излучения, вызывающего фотоэффект. Фотоэлектрическая регистрация спектров более
экспрессна, чем фотографическая. Исключается трудоемкая обработка фотопластинок
и последующие измерения почернений, соответственно устраняются погрешности,
возникающие на этих стадиях анализа. Приборы для фотоэлектрической регистрации
спектров весьма разнообразны. Качественный
анализ. Существуют обширные спектральные атласы, где приведены эталонные
спектры испускания большого числа элементов с указанием длин волн
и относительных интенсивностей линий. Однако не все линии эталонного
спектра можно найти в спектре пробы, содержащей данный элемент. По мере уменьшения
концентрации компонента в пробе интенсивность излучения уменьшается настолько,
что часть линий (наименее интенсивные)
уже не регистрируется данным прибором. Последними при разбавлении пробы
исчезают так называемые последние линии (как
правило, наиболее интенсивные). Для
каждого элемента эти линии хорошо известны. Чтобы проверить наличие некоторого
элемента в пробе, проверяют наличие в спектре пробы нескольких последних линий
этого элемента. Но если в спектре
обнаружена линия, длина волны которой численно совпадает с длиной волны последней линии искомого элемента, то
это вовсе не означает, что она действительно принадлежит данному элементу. Дело
в том, что в спектрах многих элементов насчитывается очень большое число линий
(у калия – несколько десятков, у железа – несколько сот, у урана – несколько
тысяч). Линии разных элементов часто случайно совпадают по длине волны
(«межэталонные наложения»). Могут
совпадать и последние линии.
Поэтому окончательный вывод о присутствии
в пробе интересующего элемента следует делать, если в спектре пробы установлено
наличие нескольких линий, совпадающих по длине волны с линиями эталонного
спектра данного элемента и заведомо
свободных от межэталонных наложений. Полезно также проверить наличие характерных комбинаций линий
(дублетов, триплетов). Спектры пробы и эталона должны совпадать и по относительной интенсивности разных линий.
Количественный анализ. Интенсивность
излучения в АЭС определяется концентрацией возбужденных атомов. Если все
условия анализа одинаковы, получим прямо пропорциональную зависимость
интенсивности излучения на данной длине волны
от концентрации элемента в
пробе:
![]()
Расчет концентраций
ведут по предварительно построенным градуировочным графикам.
Молекулярно-абсорбционная спектроскопия (фотометрический анализ)
Принцип
метода
Еще в начале XIX века концентрацию окрашенных растворов
научились оценивать, сравнивая на глаз интенсивность их окраски с заранее
приготовленной шкалой эталонных растворов (колориметрия).
Затем были изобретены приборы для количественного измерения поглощения света
растворами; установлены закономерности, связывающие характеристики светопоглощения
с концентрацией окрашенных веществ. В XX веке подобным образом стали определять
и концентрацию бесцветных растворов, их поглощение измеряли в УФ- или в
ИК-области. В развитие молекулярно-абсорбционной спектроскопии большой вклад
внесли физики П.Бугер (Франция),
К.Фирордт (Германия), У.Кобленц (США). В зависимости от того, в какой
области спектра измеряют аналитический сигнал, методы молекулярно-абсорбционной
спектроскопии разделяют на две группы: 1) фотометрический анализ в УФ- и
видимой области (спектрофотометрия);
2) ИК-спектроскопия. Соответствующие
методы сильно различаются по своим
возможностям, но основаны они на одних и тех же теоретических закономерностях.
Общие
закономерности поглощения света. При пропускании монохроматического светового потока через
кювету с раствором, содержащим молекулы или ионы Х, интенсивность светового
потока уменьшается. Это связано с рядом причин:
часть света поглощается молекулами или ионами Х, другая часть –
растворителем и примесями, третья - рассеивается и отражается стенками кюветы. Чтобы
учесть потери света, связанные с растворителем и кюветой, измерения проводят
относительно раствора сравнения, не содержащего Х. Обычно в качестве раствора
сравнения используют чистый растворитель. Если поместить и фотометрируемый
раствор, и раствор сравнения в одинаковые кюветы, а затем через эти кюветы
пропускать свет с одной и той же длиной
волны l и одинаковой начальной интенсивностью Iнач, то потери света на
отражение и рассеяние для обеих кювет окажутся одинаковы. Тогда различие в
интенсивности получаемых световых потоков (I
и I0) будет
определяться лишь природой и концентрацией
Х.
В качестве аналитического сигнала в
молекулярно-абсорбционной спектроскопии используют оптическую плотность (А).
Это десятичный логарифм отношения интенсивности монохроматического света,
прошедшего через раствор сравнения, к интенсивности света, прошедшего через
исследуемый раствор
![]() (1)
(1)
Оптическая плотность – безразмерная
величина. Она не зависит от Iнач,
а определяется природой и концентрацией частиц, поглощающих свет на данной
длине волны, а также толщиной поглощающего слоя в кювете. Связь этих величин
описывает основной закон светопоглощения, который принято называть законом Бугера – Ламберта - Бера:
В соответствии с этим законом, оптическая плотность
раствора, измеренная на некоторой длине волны, прямо пропорциональна
концентрации растворенного вещества,
поглощающего свет на этой длине волны, и толщине слоя раствора.
А =
ε l с (2)
Концентрацию
поглощающих частиц (С) выражают в
моль/л, толщину слоя ( l ) - в сантиметрах. В таком случае коэффициент
пропорциональности e называют молярным коэффициентом поглощения. Его
величина зависит от природы Х и длины волны, на которой измеряют оптическую
плотность.
Аппаратура. Для измерения оптической плотности
растворов и регистрации спектров поглощения используют спектрофотометры (рис.3).
Важнейшая их часть - монохроматор. Другие узлы - источник света, приемник излучения и
регистрирующее устройство.
Источники
света. В зависимости от оптической области, в которой работает прибор,
источниками света служат: в УФ-области – водородная или дейтериевая
газоразрядные лампы, дающие сплошной спектр излучения; в видимой области – обычная лампа накаливания
с вольфрамовой нитью, в ИК-области – глобар. Это керамический стержень,
нагреваемый до температур порядка 1600 0С.
Монохроматоры. В спектрофотометрах применяют
призменные монохроматоры или дифракционные решетки. Материал, из которого
изготавливают оптическую систему прибора, должен хорошо пропускать свет в
рабочем диапазоне длин волн. В УФ-области используют кварц, в видимой области –
стекло, в ИК-области – кристаллические соли, галогениды щелочных и
щелочноземельных металлов (NaCl, KBr, CaF2 ).
λ
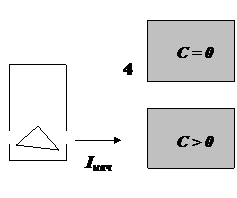

![]()
![]()
3 2
Рис. 3.
Принципиальная схема однолучевого
спектрофотометра
1 – источник света; 2 – призменный
монохроматор; 3 – кювета с исследуемым раствором;
4 – кювета с раствором сравнения; 5 –
приемник света (фотоэлемент); 6 – микроамперметр
Прибор настраивают на нулевую оптическую плотность по кювете
с раствором сравнения (4), а затем вместо нее вводят в световой поток кювету с
исследуемым раствором (3). В результате меняется интенсивность светового
потока, падающего на приемник излучения (фотоэлемент), меняется и величина
фототока.
Фотометрический
анализ
Чтобы выбрать оптимальные условия анализа, после проведения фотометрической
реакции исследуют спектр поглощения полученного соединения. Вид конкретного
спектра и значения вышеперечисленных характеристик определяются природой
поглощающих частиц. Спектр поглощения отдельного раствора строят в координатах А -
l. При изменении концентрации раствора спектральная
кривая будет сдвигаться по вертикали (рис.4, слева), но число максимумов на
этой кривой и их положение в шкале длин волн не изменятся. Длину волны, при
которой наблюдается максимальное поглощение, обозначают как lmax
, а молярный
коэффициент на этой длине волны – как εmax. Зависимость ε
от l (или lg ε от l)
характеризует все растворы данного состава. Она не меняется при изменении
концентрации растворенного вещества или толщины поглощающего слоя. Именно такие
«обобщенные» спектры поглощения индивидуальных веществ приводят в спектральных
атласах (рис.4, справа). Чем больше εmax,
тем меньшие концентрации Х можно определять по данной методике.
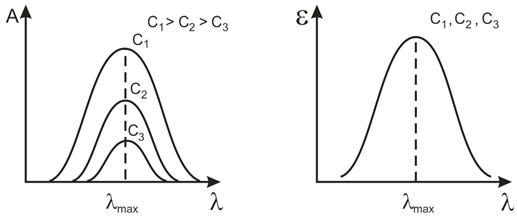
Рис.4..
Спектры поглощения растворов с
разной концентрацией Х и их обобщение
Выбор аналитической длины волны. Если проба содержит только один
компонент, поглощающий свет, то в качестве аналитической длины
волны выбирают lmax, что обеспечивает максимальную
чувствительность. Если же в растворе надо определять два и более компонента по
отдельности, аналитические длины волн выбирают так, чтобы на каждой поглощал бы
лишь один компонент. Это не всегда удается: в молекулярных спектрах полосы
поглощения достаточно широки и часто накладываются друг на друга. В таких
случаях селективность фотометрического анализа обеспечивают, проводя
соответствующую пробоподготовку. Например, маскируют или заранее отделяют один
из компонентов.
Аналитические
возможности. Простота оборудования и самих фотометрических измерений
сделали спектрофотометрию одним из самых
распространенных методов анализа. Этот метод, в отличие от АЭС, не требует
применения высоких температур, высокой квалификации исполнителей, его область
применения не ограничена задачами элементного анализа. Сегодня фотометрическим
методом аналитики решают задачи и элементного, и молекулярного, и вещественного,
и структурно-группового анализа. Для идентификации веществ этот метод применяют
редко; спектрофотометрия – это, прежде всего, способ количественного анализа.
Нижняя граница определяемых концентраций обычно характеризуется значениями
порядка 0,1 – 1,0 мкг/мл, что в большинстве случаев полностью удовлетворяет
требованиям практики. Относительная погрешность результата анализа (при
традиционном способе фотометрических измерений) составляет 2-5%, а в некоторых
случаях может быть снижена до 1%.
Весьма важно, что определяемый
компонент пробы можно заранее связать с подходящим реагентом в новое,
интенсивно окрашенное соединение. Такой прием повышает селективность анализа,
позволяет определять почти все элементы и множество их соединений. К
недостаткам фотометрического анализа можно отнести лишь невысокую селективность
и необходимость предварительного перевода
пробы в раствор.
Фотометрический анализ широко применяют в контрольно-аналитических
лабораториях на предприятиях химической, пищевой, нефтеперерабатывающей
промышленности, в криминалистике, в сельском хозяйстве, в клиническом анализе и
научных исследованиях. Особенно важен данный метод для контроля за выбросами
токсичных веществ и для мониторинга состояния окружающей среды.
Люминесцентный анализ
Принцип метода и области его применения. В определенных условиях часть поглощенной
веществом энергии может выделиться в виде вторичного излучения. Это явление
называют люминесценцией. Кванты
вторичного излучения, испускаемого люминесцирующими атомами, молекулами
или ионами, имеют меньшую энергию, чем
кванты, которые те же частицы поглощали при своем возбуждении. Виды люминесценции классифицируют по способу
возбуждения. Наиболее известны:
1.
Катодолюминесценция (свечение под действием потока
электронов, например, свечение экрана кинескопа или жидкокристаллического
экрана, свечение ламп дневного света).
2.
Хемилюминесценция (свечение за счет химической
реакции, например, у светлячков).
3.
Фотолюминесценция. В этом случае проба светится за
счет облучения невидимым УФ-светом от
внешнего источника. Именно этот вид люминесценции обычно применяют в химическом
анализе.
Виды люминесценции
классифицируют и по времени жизни возбужденного состояния. В анализе
преимущественно используют флуоресценцию – в этом случае возбужденное
состояние молекулы неустойчиво, и вторичное излучение пробы в УФ- или видимой
области спектра прекращается сразу после удаления источника возбуждения. Иногда
используют и фосфоресценцию – в этом случае свечение
вещества продолжается в течение нескольких секунд, минут или даже часов после
прекращения возбуждения. Явление люминесценции известно с давних
времен, но физики стали изучать его лишь во второй половине XIX века. В
Первые методики люминесцентного
анализа были созданы в 30-х годах XX века, во многом благодаря работам
академика С.И.Вавилова и его учеников. Сегодня
этот метод используют не очень
широко, но в некоторых областях он просто незаменим. По спектрам люминесценции
опознают особо опасные органические вещества в объектах окружающей среды (на
уровне 10-6 % и ниже). В
частности, так находят содержание
полициклических ароматических углеводородов (ПАУ), многие из которых являются
канцерогенами. Эти вещества определяют в природных и сточных водах, воздухе, почвах, продуктах питания. Тот же
метод используют для обнаружения других токсикантов (диоксины, нитрозамины,
пестициды), а также многих биологически активных веществ (витамины, гормоны,
антибиотики). Люминесцентные детекторы применяют в хроматографическом анализе,
измеряя интенсивность свечения веществ, по очереди выходящих из хроматографической
колонки. Люминесцентный анализ применяют в криминалистической экспертизе и для
диагностики заболеваний.
Люминесцировать могут далеко не все
молекулы, поглощающие свет. Большая часть органических веществ, способных
люминесцировать, – это ароматические соединения, имеющие жесткую структуру
молекулы.
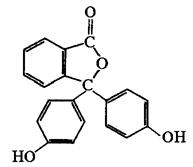
Фенолфталеин Флуоресцеин
Так, фенолфталеин и флуоресцеин имеют сходное строение молекул (см. схему).
Но у фенолфталеина все три бензольных
кольца могут свободно колебаться друг относительно друга, и это соединение не
люминесцирует. У флуоресцеина же возможность внутримолекулярных колебаний
гораздо меньше (кислородный мостик жестко фиксирует два бензольных кольца), и
это соединение интенсивно светится в видимой области при облучении его раствора
УФ-светом.
.
Аппаратура для
люминесцентного анализа. Для
идентификации индивидуальных соединений и для выбора оптимальных условий
измерения их аналитического сигнала надо изучать спектры возбуждения и спектры
испускания люминесценции. С этой целью используют различные спектрофлуориметры.
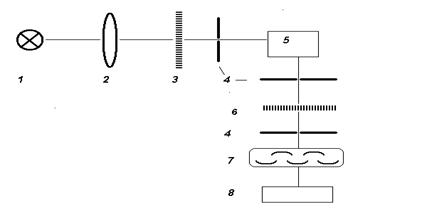
Рис.5. Принципиальная схема спектрофлуориметра
Это более сложные и дорогие приборы, чем спектрофотометры. Принципиальная схема спектрофлуориметра включает: источник возбуждающего света (1),
фокусирующую линзу (2), первичный монохроматор (3), входные и выходные щели (4), кюветное
отделение (5), вторичный монохроматор
(6), приемник люминесцентного изучения
(7), регистрирующее устройство (8).
В качестве источников возбуждения чаще всего используют мощные УФ-лампы
(ртутные, ксеноновые и др.), а также лазеры. Первичные и вторичные
монохроматоры включают дифракционные решетки или призмы, изготовленные из
кварца, а также щели регулируемой ширины. Длину волны излучения, выходящего из
первичного или вторичного монохроматора (соответственно l1 или l2), можно менять. Первичный монохроматор нужен, чтобы задать оптимальные
условия возбуждения определяемого соединения
(Х), а также задержать свет лампы с той же длиной волны, что будет испускать Х
при люминесценции. Вторичный монохроматор нужен, чтобы создать оптимальные
условия регистрации аналитического сигнала Х и не допустить попадания
возбуждающего света на фотоприемник. Без этих монохроматоров фоновый фототок
оказался бы настолько сильным, что зафиксировать аналитический сигнал Х
(люминесцентное излучение) было бы невозможно.
В упрощенных и дешевых приборах для
люминесцентного анализа (флуориметрах) вместо монохроматоров используют сменные
светофильтры (первичный и вторичный). Приемником люминесценции, как и в
спектрофлуориметрах, обычно служат фотоэлемент или фотоумножитель. Фототок усиливают, а затем измеряют с помощью
микроамперметра.
Спектры люминесценции. В обычных
условиях любые спектры люминесценции индивидуальных веществ являются
широкополосными.. Спектры испускания люминесценции похожи на спектры поглощения соответствующих
молекул. Однако возбужденные молекулы Х успевают растратить некоторую часть
ранее поглощенной энергии (на так называемые безызлучательные переходы,),
прежде чем испустят кванты вторичного излучения. Именно поэтому спектр
испускания люминесценции смещен по отношению к спектру поглощения в более
длинноволновую область. Чаще всего вещество поглощает возбуждающий свет в
УФ-области, а люминесцирует – в видимой..
Если вещество способно и
флуоресцировать, и фосфоресцировать, то его спектр испускания фосфоресценции
сдвинут в длинноволновую сторону еще сильнее, чем спектр флуоресценции (рис.6.).
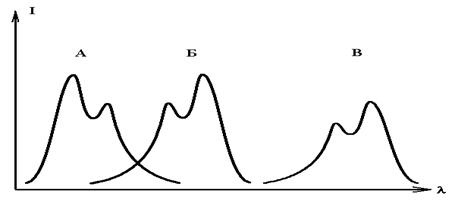
Рис 6. Спектры поглощения (А),
флуоресценции (Б) и фосфоресценции (В)
одного соединения
Качественный люминесцентный
анализ. По появлению люминесценции при
УФ-облучении пробы, а также по характерному
цвету люминесценции можно визуально опознавать некоторые элементы,
например, уран, находящийся в растворе в виде ионов. Известен целый ряд
качественных реакций, приводящих к образованию люминесцирующих соединений. Так,
например, можно открывать (а затем и определять) субмикрограммовые количества
ионов кадмия по реакции с кальцеином,
ионы бериллия и циркония – по реакции с морином, ионы цинка – с
салициловой кислотой, ионы алюминия - с
8-оксихинолином.
По
характерному цвету люминесценции выявляют и присутствие некоторых органических
соединений. Например, полиароматические углеводороды (ПАУ) дают синее свечение,
а смолы и асфальтены –
сине-зеленое. В этих случаях
люминесценцию наблюдают визуально, облучая анализируемый образец
ультрафиолетовым светом ртутной лампы. Нелюминесцирующие соединения при этом не
мешают. Чтобы выявить присутствие одного
люминесцирующего соединения в присутствии другого, придется снимать спектр
возбуждения или испускания исследуемой пробы и сопоставлять их с атласом соответствующих спектров. Но спектры
люминесценции структурно-родственных соединений (например, спектры разных ПАУ)
весьма близки. Идентифицировать индивидуальные соединения в их неразделенной
смеси по обычным (широкополосным) спектрам люминесценции удается крайне редко, спектры разных веществ
накладываются друг на друга.
Аналитические возможности люминесцентного анализа существенно расширило
одно открытие советских физиков. В
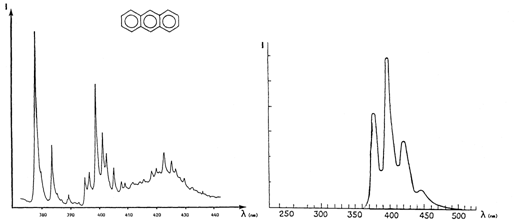
Рис.7. Спектр люминесценции раствора антрацена в н-гексане при комнатной
температуре (справа) и при -1960
С (слева)
Метрологические характеристики
люминесцентного анализа. Мы уже отмечали основное достоинство люминесцентного анализа – его высокую
чувствительность. Так можно
определять концентрации порядка 10-7
– 10-8 г/л, а в отдельных случаях – до 10-12 г/л. Весьма
высока и селективность определения люминесцирующих веществ (особенно в условиях
эффекта Шпольского). Именно поэтому люминесцентный
анализ преимущественно используют там,
где нужны высокая чувствительность и селективность, например, при оценке
загрязнения окружающей среды.