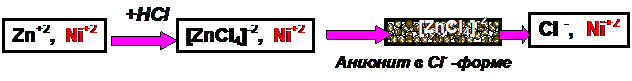Методы
разделения и концентрирования
Назначение
и классификация методов
При
выполнении химических анализов аналитики применяют множество вспомогательных
методов, нацеленных на то, чтобы на стадии пробоподготовки отделить одни
компоненты пробы от других или повысить концентрацию некоторых компонентов.
Обойтись без соответствующих операций заманчиво (каждая операция – это дополнительные
затраты времени и труда, а иногда и дополнительные погрешности), но исключить
их аналитикам удается лишь в редких случаях.
Концентрирование применяют, если намеченные способы измерения
аналитического сигнала недостаточно чувствительны и не позволяют определить
компонент в исследуемом веществе на
соответствующем концентрационном уровне. Например, необходимо точно
определять содержание канцерогенного углеводорода 3,4-бензпирена (БП) в
природной воде, где его предполагаемое содержание находится на уровне 10-9
- 10-8 г/л.
Определять концентрацию БП можно
фотометрическим методом, но при столь низком содержании оптическая плотность
раствора будет близка к нулю. Чтобы измерение оптической плотности было
достаточно точным, концентрация БП
должна быть выше 10-6 г/л, а еще лучше – выше 10-5 г/л.
Очевидно, для фотометрического
определения БП в воде надо заранее
сконцентрировать его, а именно - повысить концентрацию на 3-4 порядка.
Разделение
применяют, если имеющиеся в
распоряжении аналитика способы измерения
аналитического сигнала недостаточно селективны, то есть
сигнал определяемого компонента будет искажен другими компонентами той же
пробы. Так, при фотометрическом
определении 3,4-бензпирена оптическая плотность раствора пробы на выбранной
длине волны создается не только этим веществом, но и другими ароматическими
углеводородами, присутствующими в анализируемой воде. Чтобы получить правильный
результат анализа, надо перед измерением отделить БП от других ароматических
углеводородов.
Таким образом, методы разделения и
методы концентрирования направлены на достижение разных целей. Но реализуются они однотипными способами, поэтому в
курсе аналитической химии обычно их рассматривают совместно. Аналитики ведут
пробоподготовку так, чтобы, по возможности, одновременно и отделить
определяемый компонент (Х) от мешающих веществ, и сконцентрировать его. Кроме того, пробоподготовка должна предотвратить
ошибки, связанные с неравномерным распределением Х в
анализируемом материале.
Известно более двух десятков методов разделения и/или концентрирования. Основной группой являются равновесные методы, основанные на распределении Х между двумя фазами. Установление межфазного равновесия ведет к переходу основной части Х в исходной (первой) в новую (вторую) фазу. Приведем классификацию данных методов по агрегатному состоянию фаз в виде таблицы. Здесь и далее примем, что 1-ая фаза – это фаза, из которой следует извлечь Х, а 2-ая фаза – та, в которую Х извлекается.
Некоторые методы разделения и
концентрирования микропримесей
|
Первая фаза |
Вторая фаза |
Метод |
Основное применение в анализе |
|
Жидкая |
Жидкая |
Экстракция |
Универсальный метод |
|
Электролиз с ртутным катодом |
Концентрирование тяжелых металлов |
||
|
Жидкая |
Газообразная |
Отгонка, ректификация, дистилляция |
Разделение летучих органических веществ |
|
Упаривание пробы |
Концентрирование нелетучих веществ |
||
|
Жидкая |
Твердая |
Сорбция |
Выделение и концентрирование растворенных веществ |
|
Ионный обмен |
Разделение и концентрирование ионов |
||
|
Жидкостная хроматография (ВЭЖХ, ТСХ) |
Разделение нефтепродуктов, красителей, аминокислот, лекарственных
препаратов |
||
|
Газообразная |
Жидкая |
Газожидкостная хроматография (ГЖХ) |
Разделение органических веществ, анализ нефтепродуктов |
|
Улавливание жидкостными поглотителями |
Анализ воздуха |
||
|
Газообразная |
Твердая |
Газоадсорбционная хроматография (ГАХ) |
Анализ воздуха и других смесей легких
газообразных вешеств, определение воды |
|
Фильтрация |
Концентрирование аэрозолей |
||
|
Твердая |
Жидкая |
Селективное растворение (выщелачивание) |
Разделение компонентов почв, горных пород, лекарственных препаратов и т.п. |
|
Зонная плавка |
Концентрирование примесей в металлах и других веществах высокой чистоты |
||
|
Твердая |
Газообразная |
Озоление |
Концентрирование нелетучих примесей в биообъектах, пищевых продуктах,
нефти |
Так, сконцентрировать 3,4-бензпирен
и отделить его от мешающих веществ можно
экстракционным методом. А
именно, к большому объему исследуемой воды добавляют немного не смешивающегося
с водой органического растворителя (например, н-гексана), который хорошо
растворяет БП. После установления межфазного равновесия БП почти полностью
экстрагируется, то есть перейдет в фазу органического растворителя. Полученный раствор (экстракт)
отделяют от водной фазы и затем фотометрируют. Отметим, что в этом примере
межфазное равновесие и соответствующее ему распределение Х устанавливалось
только один раз. Более эффективны методы разделения и концентрирования, в которых происходит многократное
перераспределение каждого компонента пробы между двумя фазами. Это ведет к
полному разделению компонентов и повышает точность анализа.
Разные методы дополняют друг друга. Одни из них направлены на абсолютное концентрирование всех микропримесей без изменения соотношения их концентраций. Так, при определении тяжелых металлов в морской воде пробу упаривают, ионы всех металлов остаются в концентрированном растворе (рассоле), который затем и анализируют. Другие методы (соосаждение) направлены на относительное концентрирование некоторых микропримесей. Третьи используют лишь для разделения, а повышения концентрации микропримесей они не обеспечивают. Это характерно для многих вариантов хроматографии, хотя ее следует считать не просто одним из методов разделения, а гибридным методом. Есть универсальные методы, которые с успехом используются и для разделения, и для абсолютного, и для относительного концентрирования. Примером может быть экстракция
Количественные
характеристики процессов разделения и концентрирования
При установившемся межфазном
равновесии химические потенциалы вещества Х, распределяемого между двумя
фазами, равны, а отношение активностей Х в этих фазах - постоянная величина,
которую называют константой распределения:
КD = ![]() (7.1)
(7.1)
Если в одной из
фаз вещество Х находится в нескольких переходящих друг в друга формах,
(например, отличающихся по протонированности, закомплексованности или степени
окисления), то извлечение каждой из них в новую фазу следует характеризовать
своей константой распределения. Такие константы
можно вычислить методами химической термодинамики. Для многих межфазных
равновесий они определены опытным путем,
значения КD можно
найти в справочниках. Они не зависят от
начальной концентрации Х и не меняются в присутствии посторонних
веществ. Чем больше КD, тем лучше
извлекается Х в новую фазу.
Отношение суммарных концентраций всех равновесных форм
Х - приблизительно постоянная величина, называемая коэффициентом распределения[1]:
D = ![]() (7.2)
(7.2)
В формуле (7.2) символом ![]() 2 обозначена суммарная концентрация всех форм Х в
новой фазе, а символом
2 обозначена суммарная концентрация всех форм Х в
новой фазе, а символом ![]() 1 - суммарная концентрация всех форм Х в исходной
фазе (при установившемся межфазном равновесии). Величину D определяют
опытным путем, разделяя фазы и измеряя в них концентрации Х. Значения КD
и D взаимосвязаны. Если вещество Х может находиться
только в одной форме, тогда КD ≈ D.
Коэффициенты распределения (как и константы распределения), зависят от природы
Х, природы фаз и температуры. Кроме того, значения коэффициентов распределения
могут зависеть от присутствия посторонних веществ, а также от других факторов.
Именно это отличает D от КD. Фактически
D – это условная константа экстракционного
равновесия.
1 - суммарная концентрация всех форм Х в исходной
фазе (при установившемся межфазном равновесии). Величину D определяют
опытным путем, разделяя фазы и измеряя в них концентрации Х. Значения КD
и D взаимосвязаны. Если вещество Х может находиться
только в одной форме, тогда КD ≈ D.
Коэффициенты распределения (как и константы распределения), зависят от природы
Х, природы фаз и температуры. Кроме того, значения коэффициентов распределения
могут зависеть от присутствия посторонних веществ, а также от других факторов.
Именно это отличает D от КD. Фактически
D – это условная константа экстракционного
равновесия.
Коэффициенты распределения имеют большее практическое значение, чем
константы. В частности, зная эти
коэффициенты, аналитик может предвидеть
полноту извлечения разных компонентов пробы, возможность их разделения в
заданных условиях, и т.п. В ходе таких
расчетов используют следующие характеристики:
Степень извлечения (R) - отношение
количества вещества Х, перешедшего в новую фазу (далее обозначается как νX2), к
исходному количеству Х (νX0). Вместо
отношения числа молей можно взять отношение масс:
R = ![]() (7.3)
(7.3)
Проводя
разделение и концентрирование, стремятся, чтобы
величина R для определяемого
компонента была бы как можно ближе к 1. Разность (1 – R)
характеризует потери Х в ходе пробоподготовки. Отметим, что безразмерную
величину R выражают и
в процентах.
Коэффициент разделения (χ) двух компонентов пробы (Х и X*) позволяет заранее оценить возможность их разделения
по данной методике. Величина χ равна
отношению коэффициентов распределения: χ = Dх / Dх* . Чем сильнее отличается χ от
единицы, тем лучше будет идти процесс разделения Х и X* . Однако при этом
должно выполняться и дополнительное условие: произведение
Dх.Dх* должно быть по возможности ближе к единице. Так, если
Dх = 103, а Dх* = 10-3 ,
коэффициент разделения χ =106.
Тогда Х извлекается, а Х* практически не
извлекается. Если же Dх = 109, а Dх* = 103, коэффициент разделения имеет ту же величину,
что и в предыдущем случае, но извлекаются оба компонента, разделения нет.
Коэффициент концентрирования (N) равен отношению концентрации Х в
новой фазе (в концентрате) к исходной концентрации Х в пробе. Если потерь Х в
ходе его извлечения не было, то коэффициент концентрирования приблизительно
равен отношению объема пробы (V1) к объему концентрата
(V2), или
отношению соответствующих масс:
N
= ![]() (7.4)
(7.4)
Экстракция в
анализе
Экстракция – процесс распределения
растворенного вещества (Х) между двумя несмешивающимися жидкими фазами,
используемый для перевода Х из одной фазы в другую. В
аналитических лабораториях экстракцию применяют для индивидуального и
группового концентрирования микропримесей, для отделения мешающих
макрокомпонентов пробы («сброс матрицы»), а также для очистки реактивов и
растворителей. Отметим, что экстракционные методы используются не только в
анализе, но и в технике, в частности, в химической, нефтеперерабатывающей,
фармацевтической и пищевой промышленности. Экстракционные процессы лежат в
основе химической чистки одежды, они широко применяются в технологии редких
металлов и особо чистых веществ. Но в данном учебнике будет рассматриваться
только применение экстракции в анализе. Этот способ пробоподготовки известен с
конца XIX века, но широко применять
его стали лишь в 50-х годах XX века.
Экстракционные методы определения микропримесей разработаны в значительной
степени благодаря исследованиям отечественных аналитиков (Ю.А.Золотов,
Я.И.Коренман и др.).
Как метод
пробоподготовки, экстракция имеет несомненные преимущества, а именно:
·
универсальность. Соответствующие методики
экстракционного извлечения разработаны практически для всех элементов и для
многих органических веществ;
·
достаточно
высокие коэффициенты концентрирования (102 – 104);
·
высокая
селективность, позволяющая разделять близкие по свойствам вещества. Селективность
можно дополнительно регулировать, меняя
рН или вводя маскирующие вещества;
·
возможность проведения процесса при комнатной
температуре и атмосферном давлении;
·
простота
методик, низкая стоимость оборудования и материалов, малые затраты времени.
Экстракция имеет и свои недостатки. В частности, она требует больших трудозатрат, плохо
поддается автоматизации, многие
экстрагенты токсичны и пожароопасны.
Техника проведения экстракции. В анализе
неорганических веществ и объектов окружающей среды определяемые вещества
экстрагируют из водного раствора в неводную фазу. В этих случаях в качестве
экстрагентов применяют несмешивающиеся с водой органические растворители[2].
Примером может быть экстракция 3,4-бензпирена н-гексаном из природных вод. В анализе органических веществ
экстрагентом может быть и вода. Так, исследуя состав нефтепродуктов или
растительных масел, экстрагируют соли и другие полярные компоненты пробы
дистиллированной водой или водным раствором кислоты.
Экстрагирование можно вести разными
способами:
·
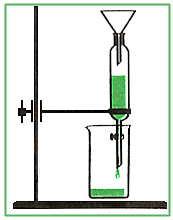 однократная экстракция в
делительной воронке, куда вводят большой объем раствора пробы и немного
экстрагента. Воронку встряхивают вручную в течение определенного времени, после
прекращения встряхивания смесь расслаивается и нижнюю фазу сливают через кран.
Затем экстракт анализируют;
однократная экстракция в
делительной воронке, куда вводят большой объем раствора пробы и немного
экстрагента. Воронку встряхивают вручную в течение определенного времени, после
прекращения встряхивания смесь расслаивается и нижнюю фазу сливают через кран.
Затем экстракт анализируют;
·
периодическая
экстракция при многократном добавлении экстрагента к раствору пробы.
После ввода каждой порции воронку встряхивают и отделяют экстракт. Полученные
экстракты объединяют. Такой способ позволяет количественно извлечь Х из пробы,
даже если при установлении равновесия значительная доля Х остается в водной фазе;
·
непрерывная
экстракция проводится в специальных аппаратах (экстракторах), обеспечивающих
длительный контакт фаз. Обычно экстрагент пропускают через раствор пробы до тех
пор, когда практически весь Х перейдет в экстракт. Иногда в экстракторах
устанавливают встречное движение обеих фаз (противоточная
экстракция), что еще более повышает эффективность процесса. В
экстракторах предусмотрены и устройства
для непрерывной регенерации экстрагента. В аналитических лабораториях
экстракторы используют редко, гораздо чаще их применяют в химической технологии.
Традиционные методики пробоподготовки обычно
включают 2-4 повторные экстракции. Каждый раз
берут одинаковые по объему порции одного и того же экстрагента. Условия
обработки (время контакта фаз,
интенсивность встряхивания, рН водной фазы и т.п.) также не меняют.
Объединенный экстракт анализируют каким-либо инструментальным методом. Если
использовать атомно-абсорбционный, спектрофотометрический или люминесцентный
метод, аналитический сигнал Х можно измерять прямо в экстракте. Можно также
ввести каплю экстракта в хроматограф и получить хроматограмму, на которой будут
видны пики всех веществ, извлеченных из пробы. Если же сигнал Х собираются
измерять каким-либо электрохимическим или другим методом, то придется проводить
реэкстракцию, то есть переводить
Х из экстракта в новый водный раствор.
Для этого экстракт встряхивают в делительной воронке с небольшим количеством
водного раствора сильной кислоты. Реэкстракция позволяет лучше сконцентрировать
Х и отделить мешающие вещества.
Экстракт Б Экстракт А
![]()
![]()
![]()
![]()


Рис.7.1. Схема
извлечения микропримеси Х из водного
раствора
легкими (А) и тяжелыми (Б)
экстрагентами
В
качестве экстрагентов традиционно используют жидкие при комнатной температуре
чистые органические растворители. В
состав экстрагента могут входить и другие вещества (разбавители, модифицирующие
добавки, экстракционные реагенты). Экстрагенты должны соответствовать следующим
требованиям:
·
растворимость Х в экстрагенте должна быть значительно
выше, чем в воде. Наоборот, вещества, мешающие определению Х, в экстрагенте
растворяться не должны;
·
экстрагент не должен растворять воду, а вода не должна
растворяться в экстрагенте;
·
летучесть паров экстрагента должна быть как можно
меньше. Предпочтительны экстрагенты с высокой температурой кипения;
·
плотность экстрагента
должна сильно отличаться от плотности водных растворов, иначе двухфазная
система не будет расслаиваться. В
качестве экстрагентов применяют как легкие растворители (эфиры, углеводороды),
так и тяжелые галоидсодержащие вещества, более тяжелые, чем вода (рис.7.1);
·
экстрагент должен быть дешевым, малотоксичным,
устойчивым при хранении.
Этому
комплексу требований отвечают лишь немногие органические вещества. Чаще всего
применяют тетрахлорметан (четыреххлористый углерод, CCl4), хлороформ (CHCl3), н-гексан, изооктан, изобутиловый и изоамиловый
спирты, некоторые амины, сложные эфиры. К сожалению, в лабораториях приходится
использовать и токсичные экстрагенты (бензол и т.п.). Экстракционное разделение
и концентрирование обязательно требует сильной тяги. Экстракты после завершения
анализа нельзя сливать в канализацию, их собирают и очищают для повторного
использования экстрагента. Регенерацию обычно ведут путем перегонки. В
последние годы в качестве экстрагентов стали использовать расплавы некоторых
легкоплавких веществ, например, нафталина. При небольшом охлаждении экстракт
застывает и от него легко отделяется жидкая фаза.
Часто аналитику не удается
подобрать подходящий экстрагент. Так бывает, когда определяемый компонент
хорошо растворим в исходной фазе. Другая причина - неселективность
экстрагентов, из-за чего в новую фазу одновременно переходят и Х, и родственные
ему мешающие примеси. В обоих случаях надо перевести Х в новую, хорошо
экстрагируемую форму. Для экстракции заряженных частиц в органическую фазу
их связывают с каким-либо противоионом,
добавляя заранее (или вместе с экстрагентом) подходящий реагент. Так, катионы
тяжелых металлов без добавления реагентов практически не экстрагируются из водных
растворов. Поэтому в анализе природных вод широко применяют экстракционные реагенты, связывающие
металлы в нейтральные комплексы (дитизон, оксихинолин, карбаминаты,
нитрозонафтолы и т.п.). В отличие от свободных ионов металла, такие комплексы
плохо растворимы в воде, но хорошо растворимы в органических растворителях. При
добавлении реагента коэффициент распределения металла между двумя фазами резко
повышается, и металла количественно переходит в экстракт. При определении
неметаллов в качестве экстракционных реагентов применяют органические
красители, образующие с неметаллом нейтральный
ионный ассоциат. Так, бор во фторидных растворах существует в основном в виде
аниона BF4-. В кислой
среде этот анион дает нейтральный ассоциат с катионной формой красителя
метилового фиолетового, и полученный ассоциат экстрагируют бензолом. В любом
случае экстракционный реагент подбирают так, чтобы он реагировал с Х селективно.
Экстракционное разделение микропримесей. Хотя экстракцию чаще применяют как
метод концентрирования, с ее помощью можно и разделять сложные смеси веществ, в
том числе с близкими свойствами. Для этого используют следующие способы:
1). Подбор селективного экстрагента (растворителя).
Этот способ преимущественно используется при экстракции органических веществ,
когда никакие химические реакции не происходят, а механизм экстракции сводится
к простому физическому распределению. Подбирают такой экстрагент, в котором
разделяемые вещества как можно сильнее
различаются по своей растворимости.
2) Подбор
селективного экстракционного реагента. Этот способ преимущественно используют для
экстракционного разделения
неорганических микропримесей, когда механизм экстракции включает образование хелатных соединений.
Подбирают такой реагент, который дает прочные нейтральные комплексы с определяемыми веществами, но не дает их с мешающими
веществами. Так, раствор органического реагента дитизона в хлороформе используют в анализе технической поваренной
соли для извлечения и последующего определения тяжелых металлов (свинца, меди,
кадмия и др.). Поскольку ионы натрия не дают дитизонатных комплексов, хлорид
натрия остается в водной фазе. Отметим, что комплексы дитизона с тяжелыми
металлами интенсивно окрашены, поэтому фотометрируя экстракт на подходящих
длинах волн, можно определить концентрацию отдельных металлов. Такой метод
называют экстракционно-фотометрическим.
Можно
найти и такие экстракционные реагенты, которые взаимодействуют лишь с некоторыми
переходными металлами. Например, органический реагент диметилглиоксим (реактив
Чугаева) дает прочные, хорошо экстрагируемые и интенсивно окрашенные комплексы
только с ионами никеля, палладия и железа.
3) Подбор рН. Этот способ применяют для разделения веществ,
которые различаются по своим кислотно-основным свойствам. Так, углеводороды, не
способные к реакциям протолиза, извлекаются органическими растворителями из водной
фазы при любом рН, а фенолы (слабые кислоты) в щелочной среде существуют в виде
неэкстрагируемых анионов. Проводя экстракцию в сильно щелочной среде, можно
отделить углеводороды от фенолов.
Величина
рН сильно влияет и на экстракцию комплексных соединений. Обычно в водной
фазе проходят две конкурирующие реакции:
M + n
R
↔ MRn ↔ MRn (в
экстракте)
R + H+ ↔ НR+
Вторая реакция (протонирование экстракционного
реагента) сдвигает влево равновесие первой реакции. При подкислении
раствора комплексы реагентов с металлами
разрушаются, и эти металлы перестают экстрагироваться. Но комплексы разных металлов с одним и тем же реагентом
имеют различную прочность, поэтому они
устойчивы в разных областях рН. Примером
могут быть уже упоминавшиеся комплексы металлов с дитизоном. Их прочность падает в ряду
Ag > Cu > Sn > Pb > Mn. Серебро
отделяют от других металлов, проводя экстракцию при рН 0 (в столь кислой среде
другие перечисленные металлы комплексов не образуют). Повысив рН водной фазы до
2-3 единиц, можно извлечь медь, при рН 5 – олово, а при рН 7 - свинец. Если же два металла дают
комплексы одинаковой прочности (например, серебро и ртуть), разделить их таким
способом не удается.
4) Маскирование
применяется для разделения
веществ, дающих экстрагируемые комплексы одинаковой прочности. Надо только
найти селективно действующий маскирующий реагент. Так, разделяя ионы Fe3+ и Ni2+, можно
ввести в анализируемую пробу избыток винной кислоты. Она свяжет железо, и
никель можно будет экстрагировать хлороформом в виде комплекса с
диметилглиоксимом. Без винной кислоты
экстрагировались бы оба металла.
5) Неравновесная экстракция. В некоторых
случаях экстрагирование микропримесей
ведут, не дожидаясь установления
равновесия. Этот прием полезен при разделении компонентов пробы с близкими
коэффициентами распределения. Разделение возможно, если скорости взаимодействия компонентов с экстракционным
реагентом заметно различаются. Так, в частности,
отделяют цинк, медь и многие другие переходные металлы от хрома(III). Экстрагируемые комплексные соединения хрома с
дитизоном образуются медленно, тогда как аналогичные соединения цинка и меди
образуются практически мгновенно. Поэтому при кратковременной экстракции медь и
цинк извлекаются, а хром остается в водной фазе.
В
специальной литературе описаны и другие способы экстракционного разделения веществ
(обменная экстракция, экстракционная хроматография и т.п.), но перечисленные
выше варианты используют наиболее часто.
Ионообменные процессы в анализе
Иониты. Некоторые твердые вещества способны обратимо поглощать ионы
из раствора, одновременно выделяя в него эквивалентные количества ионов того же
знака, ранее довольно прочно связанных в
структуре или на поверхности этого вещества (ионообменника). Обратимо обменивать ионы способны многие природные
вещества (минералы, почвы и даже ткани живого организма). Обменную емкость
характеризует число молей ионов, поглощаемых одним граммом (или одним
миллилитром) ионообменника за счет обмена в статических или динамических
условиях. Измерение обменной емкости проводят в строго определенных условиях
(температура, рН, скорость пропускания
раствора через ионит и т.п.). Обменная емкость природных материалов
невелика, поэтому в химическом анализе с 30-х годов XX века используют специально синтезированные ионообменные
материалы (катиониты и аниониты). Емкость ионитов намного
выше, чем у природных ионообменников. Некоторые иониты могут поглощать до 10
миллимолей ионов на
Структурной основой синтетических
ионитов обычно служат сополимеры различных производных стирола и
дивинилбензола, то есть по составу
иониты родственны синтетическим
каучукам. Соответствующие макромолекулы имеют линейную, сетчатую или, чаще,
трехмерную структуру и содержат множество функциональных групп. В катионитах содержатся кислотные
группы (-SO3H, -COOH, -OH и т.п.),
способные к обратимому обмену водорода на
другие катионы. Функциональные группы анионитов (-NH3+, = NH2+ и др.) за
счет своего положительного заряда присоединяют анион (например, ОН-),
который в дальнейшем может обмениваться на другой анион (из раствора).
Иониты, как правило, представляют
собой мелкие гранулы примерно одинакового размера[3].
В воде они нерастворимы. Обработка концентрированными растворами кислот, щелочей или солей переводит ионит в новую форму, отличающуюся природой ионов, способных
к обмену. Так, анионит можно заранее перевести с помощью раствора NaCl из OH-формы
в Сl-форму, катионит – из
Н-формы в Na-форму, и
т.д. Перед применением ионит отмывают чистой водой от реагентов, использованных
для его перевода в нужную форму, а также от примесей. Подготовленный ионит
можно просто высыпать в колбу с
анализируемым раствором, в этом случае ионный обмен пройдет в
статических условиях и завершится
установлением равновесия. Однако в аналитических лабораториях почти
всегда процессы ионного обмена проводят в динамических условиях. Для этого
ионит загружают в специальную ионообменную колонку, а затем медленно пропускают
через нее раствор пробы. За счет многократного установления равновесия процесс
обмена в колонке идет эффективнее, чем в статических условиях. Обменная емкость
ионита используется полнее, и при достаточной
длине колонки удается добиться полного поглощения Х.
Если
исследуемый раствор имеет слишком высокую концентрацию солей или если взят
слишком большой объем раствора, обменной емкости колонки не хватит, произойдет
“проскок”, то есть часть катионов пройдет в приемник, не вступив в реакцию
обмена. Проскок может произойти по еще
одной причине - если пропускать раствор через колонку слишком быстро,
равновесие ионного обмена не успевает устанавливаться. Проскок ионов приведет к
систематическим погрешностям анализа.
![]()
![]() Ионообменное равновесие. Реакции ионного обмена можно представить так:
Ионообменное равновесие. Реакции ионного обмена можно представить так:
на катионите: R-M1 + M2+ ↔
R-M2 + M1+
![]()
![]() на анионите: R-A1 + A2
- ↔ R-A2 + A1-
на анионите: R-A1 + A2
- ↔ R-A2 + A1-
Горизонтальная черта над формулой указывает на твердую фазу.
Применение ионитов. В
аналитических лабораториях применение ионитов можно считать одним из
сорбционных (точнее, хемосорбционных) методов разделения и концентрирования.
Способы применения ионитов многочисленны и разнообразны, например:
а) Получение
деионизованной воды. Чтобы очистить дистиллированную воду от микроколичеств
любых солей, можно вначале пропустить ее
через колонку с катионитом в Н-форме, затем через колонку с анионитом в
ОН-форме. В первой колонке катионы солей будут замещены на ионы водорода, во
второй – анионы солей будут замещены на гидроксил-ионы. Взаимодействие продуктов
обмена - Н+ и ОН—ионов
даст чистую (деионизованную) воду. На
такой воде в аналитических лабораториях готовят растворы, необходимые для
определения микропримесей. Этот способ очистки воды более эффективен, чем
обычная перегонка, однако примеси молекулярного характера таким способом удалить из воды нельзя.
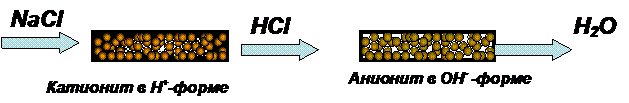
б). Отделение
электролитов от неэлектролитов. Проводится для устранения влияния мешающих
веществ. Так, рефрактометрическим методом
нельзя определить сахарозу, если в исследуемом растворе одновременно
присутствуют какие-либо соли. Поэтому
такой раствор вначале пропускают через колонку с катионитом в Н-форме, затем через колонку с анионитом в
ОН-форме. Электролиты поглощаются. Затем измеряют показатель преломления раствора,
в котором остались только молекулы сахарозы;
в)
Определение общей минерализации. Если пропустить исследуемый
раствор (например, сточную воду) через катионит в Н- форме, то соли,
содержащиеся в пробе, будут замещены эквивалентными количествами своих кислот;
раствор на выходе из колонки подкисляется:
![]()
![]() R-H
+ M+ Û R-M + H+ .
R-H
+ M+ Û R-M + H+ .
Оттитровав полученную сумму кислот стандартным
раствором щелочи, определяют суммарную концентрацию солей в исходном растворе
(общую минерализацию) в моль–экв/л.
г) Концентрирование.
Пропускают большой объем исследуемого раствора через колонку с катионитом, а
затем вытесняют (элюируют) поглощенные катионы металлов из колонки, пропуская через
нее малый объем кислоты. Концентрат анализируют подходящим инструментальным
методом, например, спектрофотометрическим или потенциометрическим.
д)
Разделение смесей ионов разного знака. С помощью ионообменных колонок
легко производится разделение компонентов пробы, образующих ионы разного знака.
Например, можно отделить никель от цинка и других металлов, обычно мешающих
определению никеля. Методика основана на том, что в солянокислой среде многие
металлы образуют анионные хлоридные комплексы, никель же таких комплексов не
дает и в солянокислом растворе находится в катионной форме. Поэтому ионы Ni2+ без
задержки пройдут через колонку с анионитом, в которой останутся анионы ZnCl4-2 и аналогичные им анионные комплексы
других металлов.