Физико-химические и физические методы анализа. Общие вопросы
. Классификация
инструментальных методов. Градуировочные функции.
Все инструментальные (физические и физико-химические) методы основаны на измерении физических величин, характеризующих объект анализа (пробу).
Измеряемая в ходе анализа физическая величина, функционально связанная
с содержанием только определяемого компонента Х в исследуемом объекте,
называется аналитическим сигналом.
Для каждого метода характерен свой аналитический сигнал. В таблице 1 приведены примеры сигналов и соответствующих им методов, относящихся к двум важнейшим группам – оптическим и электрохимическим методам анализа.
Таблица 1.
Примеры инструментальных методов анализа
|
Группа методов |
Методы (примеры) |
Аналитический сигнал |
Вид градуировочной функции |
|
|
Первичный, I |
Вторичный, I* |
|||
|
Оптические |
Атомно-эмиссионный спектральный анализ |
Фототок, i; относительное почернение, DS |
|
i = a Cb DS = a + k lg C |
|
Спектрофотометрия |
Оптическая плотность, А |
|
D = e l C |
|
|
Электрохимические |
Потенциометрия |
Э.д.с. электрохими-ческой
ячейки, DЕ
|
Потенциал электрода, Е |
Е = a + b lgC |
|
Вольтамперометрия |
Сила тока, i |
Предельный диффузионный ток, id |
Id = kC |
|
Выделяют и другие группы методов. Так, методы, в которых измеряют радиоактивность, относят к ядерно-физическим. Выделяют масс-спектрометрические методы, термические методы и т.д. Эта классификация условна и не является единственно возможной.
Зависимость аналитического сигнала от содержания Х называют градуировочной функцией. Ее записывают как уравнение вида I = f (C). В этом уравнении символом С обозначают содержание Х, выраженное в единицах количества вещества (моль), единицах массы (кг, г) или концентрации (моль/л и др.); эти величины прямо пропорциональны друг другу. Величину сигнала в общем случае обозначают символом I, хотя в отдельных методах используют специфические обозначения (см. таблицу 1). В каждом методе градуировочные функции однотипны, но точный вид градуировочной функции для конкретной методики зависит от природы Х и условий измерения сигнала.
Во многих методах зависимость сигнала от концентрации описывается нелинейными функциями, например, в люминесцентном анализе – показательной (I = kCn ), в потенциометрии - логарифмической (I = I0 + k lgC ), и т.д. Однако все градуировочные функции схожи тем, что по мере возрастания С величина I изменяется непрерывно, а каждому значению С соответствует единственное значение I.
I

![]()
![]()
![]()
![]()
Рис.1. Типичные градуировочные графики для некоторых
инструментальных методов
Градуировочные
функции устанавливают опытным путем, используя образцы сравнения (эталоны). Содержание компонента Х в каждом
эталоне заранее известно. Данные, полученные в результате измерения сигнала по
каждому эталону, позволяют представить градуировочную функцию в виде таблицы,
графика или алгебраической формулы. Если
теперь измерить сигнал пробы - на той же аппаратуре и в тех же условиях, что и
сигнал эталона, - то с помощью градуировочной функции можно будет рассчитать
содержание Х в пробе. Проще всего рассчитать результат анализа, если сигнал
прямо пропорционален содержанию Х. Если это не так, то непосредственно
измеряемый (первичный) сигнал I обычно преобразуют в новую форму I*. Можно пересчитать все значения I в I* и позднее, при
построении градуировочного графика. Способ преобразования выбирают так, чтобы
зависимость I* от содержания
Х была прямо пропорциональной и хорошо воспроизводимой. Для этого зачастую используют
логарифмирование.
Чувствительность и селективность методик
В
инструментальных методах важнейшей характеристикой любой методики анализа является
ее чувствительность (точнее называть
эту характеристику коэффициентом чувствительности). Она показывает, насколько
сильно меняется сигнал при изменении содержания Х.
К = D I
/ D C (1)
Величину K можно приблизительно оценить, разделив разность аналитических сигналов
двух однотипных эталонов на разность
содержаний Х в этих эталонах.
Для
определения одного и того же Х можно использовать множество его сигналов, и необходимо
сделать выбор заранее. Например, в атомно-эмиссионном спектре раствора NaCl имеется множество линий натрия,
отличающихся по длине волны (λ). Подготовив серию эталонов (растворов
с разной, но точно известной концентрацией NaCl), можно
измерить интенсивность излучения атомов натрия
на любой из этих длин волн. Затем строят градуировочный график, который
в узком диапазоне концентраций будет прямолинейным (рис 1). Но надо заранее
решить, на какой длине волны проводить все последующие измерения. Обычно в
спектре натрия выбирают наиболее интенсивную линию, она будет самой
чувствительной, дающей наибольшую
крутизну градуировочного графика.
I Imin щ0
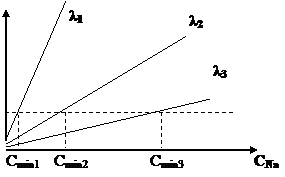
![]() Рис.2. Градуировочные графики для определения натрия на
трех разных длинах волн
Рис.2. Градуировочные графики для определения натрия на
трех разных длинах волн
Допустим, минимальное значение
сигнала, которое еще можно измерить с помощью некоторого спектрального прибора
(Imin),
одинаково для разных длин волн. Как видно из рис.1, наименьшие содержания
натрия можно будет определять, если проводить измерения на длине волны λ1, соответствующей самой интенсивной
(самой чувствительной) линии в спектре натрия. При измерении аналитических сигналов Х величину К можно менять, усиливая или ослабляя сигнал чисто аппаратурными
средствами, подобно тому как меняют громкость звучания радиоаппаратуры. Однако
такой способ применим далеко не ко всем методам.
Селективность. Условия анализа надо
оптимизировать не только по чувствительности, но и по селективности. Дело в том, что в выбранных условиях
однотипные сигналы могут одновременно создавать разные компоненты одной и той
же пробы, например вещества Х и Y. Тогда прибор зарегистрирует суммарный
сигнал:
I = Kx Сх +
KY CY (2),
и обычный способ
определения Х по градуировочному
графику, построенному без учета присутствия Y, даст завышенные результаты. Для снижения нежелательного влияния Y на величину аналитического сигнала
других компонентов пробы их предварительно маскируют или отделяют в ходе пробоподготовки.
Фон и способы его снижения. Если взять серию проб с разным
содержанием Х и измерять для них физическую величину, служащую аналитическим
сигналом, то, как правило, результат
измерений окажется ненулевым и в тех пробах, где Сх = 0.
Результат измерения аналитического сигнала
в отсутствие Х называют фоновым сигналом или просто фоном.
Например,
радиоактивность урановой руды зависит от содержания в ней урана. Однако
измерительный прибор (счетчик Гейгера, радиометр) покажет наличие некоторой
(обычно очень малой) радиоактивности в любой пробе, в том числе и в той,
которая заведомо не содержит урана. Показания прибора не будут равны нулю даже
при проведении измерений без всякой пробы! Природный радиоактивный фон
создается не ураном, а космическим излучением и микропримесями радиоактивных
изотопов, содержащихся в атмосфере. Естественно, тот же фон регистрируется и
при измерении радиоактивности урансодержащих проб, вместе с сигналом урана.
В любых физических методах
показания аналитического измерительного прибора (I) включают две составляющие –
сигнал определяемого компонента (IХ) и фон (I0). Наличие фона осложняет расчет
содержания Х. Вычислять значения IХ можно, вычитая фон из каждого результата измерений, но это
не так просто сделать: фон
нестабилен, его уровень колеблется от измерения к измерению. Поэтому аналитики
стараются не просто учесть уровень фона, но и снизить его, а еще лучше -
полностью устранить. Снижение фона особенно важно при определении микропримесей.
Источники фона. Хотя в каждом аналитическом методе существуют
свои источники фона, можно указать три
источника, характерные для всех методов:
1. Фон создается примесями определяемого компонента, которые попадают в
пробу в ходе ее отбора и в процессе пробоподготовки. Аналитики сталкиваются с этим фактором при определении веществ, широко
распространенных в природе. В пробу обычно попадают извне микрограммовые
количества воды, соединения железа, кремния, алюминия и натрия. При определении
этих веществ надо особенно тщательно следить за «химической стерильностью» всех
операций, исключить возможность
загрязнения пробы. Источниками загрязнения
могут быть недостаточно чистые реактивы и растворители, плохо вымытая посуда, лабораторный
воздух. В некоторых случаях (при определении субмикрограммовых количеств цинка,
сурьмы, кремния и т.п.) опасным источником фона может стать даже косметика на
лице исполнительницы анализа! Наоборот, при определении урана или пенициллина
вряд ли можно ожидать, что проба будет загрязнена этими веществами, редко
встречающимися в аналитической лаборатории. Случайное их попадание в пробу в ходе ее отбора и в процессе пробоподготовки
маловероятно.
Возможность загрязнения пробы определяемыми
веществами выявляют заранее, проводя контрольный («холостой») опыт и измеряя
величину I0. Высокий уровень I0 говорит о необходимости улучшить
методику анализа – например, использовать в ходе пробоподготовки реактивы
большей чистоты. Если сделать этого нельзя,
вычитают величину I0 из результатов всех последующих измерений аналитического сигнала.
2. Второй источник фона – компоненты пробы, которые создают свой сигнал в
тех же условиях, что и определяемое вещество. Например, измеренная
радиоактивность урановой руды вызывается не только ураном, но и примесью тория.
В эмиссионном спектральном анализе фон связан со случайным наложением
спектральных линий других элементов на аналитическую линию элемента Х. В
кондуктометрическом анализе растворов фон создается всеми посторонними электролитами, а также
растворителем.
Чтобы
снизить фон, связанный с влиянием посторонних веществ, можно либо заранее
отделять их от определяемого компонента, либо переходить к таким условиям
измерений, в которых влияние посторонних веществ сказывается слабее. Если эти
приемы не помогают, приходится переходить к
более селективным методам анализа.
3. Третьим источником фона является сам процесс измерения сигнала на
данном приборе. Так, в спектральном
анализе интенсивность излучения пробы регистрируют с помощью фотоэлемента,
измеряя фототок. Некоторый «темновой» ток
фиксируют и в том случае, когда проба никакого излучения не дает. Это
связано с попаданием на фотоэлемент света от других источников, а также с
работой электронных устройств, усиливающих и регистрирующих фототок. Очевидно,
для устранения третьей составляющей фона
нужно правильно настроить измерительный прибор перед началом измерений. Иногда
приходится менять режим работы или конструкцию прибора.
Предел обнаружения. Важнейшей
характеристикой любой методики анализа является предел обнаружения. Его
величину во многом определяет уровень фона.
Пределом обнаружения называют минимальное
содержание компонента в пробе,
которое можно достоверно обнаружить с помощью данной методики. Для
инструментальных методов предел обнаружения – это то содержание Х, которое
достоверно повышает аналитический сигнал
по сравнению с фоновым уровнем.
Предел обнаружения находят по
экспериментальным данным и обозначают символом Сmin. Dеличина Сmin определяется, во-первых,
чувствительностью методики, а во-вторых
– величиной минимально измеряемого
сигнала (Imin).
Аналитики заинтересованы в
снижении величины Сmin, чтобы можно было определять
микропримеси. Gредел обнаружения можно снижать
двумя путями. Во-первых, нужно увеличивать чувствительность методики
(величину К). Для этого из множества возможных сигналов Х выбирают наиболее
интенсивный (рис.2), оптимизируют условия его измерения, используют
высокочувствительные приборы, дополнительно усиливают сигнал и т.п. Во-вторых,
нужно улучшать воспроизводимость измерений . Если снизить фон не удается, его
следует хотя бы стабилизировать, а для этого проводят все измерения на одной и
той же аппаратуре и в строго одинаковых условиях. Перед началом измерений дают
прибору прогреться или термостатируют его, включают приборы в сеть через стабилизаторы
напряжения, и т.п. Эти приемы снижают Сmin даже при неизменном среднем уровне
фона.
Предел обнаружения характеризует
конкретную методику, то есть процесс определения некоторого Х с помощью данного
прибора в данных условиях. Этот предел
зависит и от природы Х, и от состава пробы, и от других факторов. Тем не
менее, в рамках группы однотипных методик пределы обнаружения разных Х
относительно близки (таблица 1).. Так, атмоно-эмиссионные методики определения
любых Х имеют довольно высокие значения Сmin (малочувствительны), а методики,
основанные на измерении люминесценции, -
высокочувствительны, то есть имеют низкие пределы обнаружения (10-8
– 10-13 М).
Таблица 1.
Пределы обнаружения, характерные для некоторых аналитических методов
|
Метод |
- lg Cmin |
||
|
Атомно-эмиссионный спектральный анализ (пламя) |
3-5 |
|
|
|
Полярография |
5-6 |
|
|
|
Потенциометрия |
5-7 |
|
|
|
Атомная абсорбция (пламя) |
5-7 |
|
|
|
Спектрофотометрия |
5-8 |
|
|
|
Инверсионная вольтамперометрия |
8-10 |
|
|
|
Люминесценция |
8-13 |
|
|
Инструментальные
методы качественного анализа
Большинство инструментальных методов селективно, их можно использовать
как для количественного, так и для качественного анализа. Применение
инструментальных методов в настоящее время стало основным способом
качественного анализа; эти методы используются гораздо чаще, чем химические
методы обнаружения ионов. При проведении качественного анализа целью является отнесение исследуемого материала к
какому-то типу объектов и/или идентификация всех или некоторых компонентов
этого материала. Компонент называют идентифицированным,
если его присутствие в пробе доказано с
требуемой надежностью. В ходе качественного анализа приходится решать
идентификационные задачи разного типа. А именно:
·
подтверждение природы чистого
вещества (проверка предполагаемого состава однокомпонентной пробы),
·
установление состава чистого вещества
(однокомпонентной пробы),
·
проверка присутствия некоторых
предполагаемых компонентов в пробе сложного состава,
·
полный качественный анализ
(выявление всех компонентов смеси неизвестного состава),
·
опознание неизвестного вещества
сложного состава как целостного объекта.
Сложность
задач в этом ряду нарастает, а надежность получаемых ответов падает. Смотря по
тому, какую задачу решает аналитик, меняются и способы проведения качественного
анализа. В частности, он может применять те или иные инструментальные методы.
Подтверждение природы чистого вещества.
Для решения этой задачи сложные и высокочувствительные
инструментальные методы использовать не обязательно, но необходимо заранее
проверить, действительно ли проба является чистым веществом. С этой целью обычно используют хроматографические методы. Другой прием – измерение физических свойств
пробы: температуры кипения, плотности, показателя преломления и т.п., Если
проба кипит или плавится не при определенной температуре, а в некотором интервале температур, она
является не чистым индивидуальным веществом, а смесью. Если же проверка
показала, что проба является чистым веществом, сопоставляют результаты
проведенных измерений с аналогичными характеристиками предполагаемого
соединения Х, имеющегося в лаборатории в чистом виде и используемого в качестве
образца сравнения (ОС, эталона). Вероятно, лучшим способом подтверждения
состава пробы является сопоставление спектров поглощения (или испускания) пробы
и эталона, например, в инфракрасной области. Чем больше признаков Х
использовано для проверки, чем больше выявлено совпадающих характеристик пробы
и эталона, - тем более надежной будет идентификация Х. Если проба действительно
является веществом Х, все ее
характеристики с точностью до
погрешности измерений должны совпасть с
непосредственно измеренными или табличными характеристиками вещества Х.
Отметим, что применение табличных (справочных) данных облегчает проверку, но
несколько снижает надежность ее результатов.
Важно правильно сформулировать выводы. Если
хотя бы одна характеристика пробы окажется достоверно отличающейся от
соответствующей характеристики ОС, проводят дополнительную проверку, а
обнаружив дальнейшие несовпадения, делают категорический вывод: «Данная проба не является веществом Х».
Если же выявленные расхождения характеристик пробы и ОС не превышают
погрешностей измерения или таких расхождений вообще нет – вывод должен
быть осторожным: «Достоверные различия пробы и вещества Х не выявлены. Можно считать, что
проба представляет собой вещество Х”. Характер вывода объясняется тем, что
обнаруженные совпадения пробы и эталона могли быть и случайными! Возможно, проверка
по другим, более специфическим признакам Х показала бы, что на самом деле
исследуемая проба содержит не вещество Х,
а лишь похожее на него соединение.
Опознание чистого вещества. Эта задача
похожа на предыдущую, но гораздо труднее. Поскольку аналитик не имеет
обоснованных исходных предположений о составе пробы, ему пришлось бы сравнивать физические свойства пробы с
аналогичными характеристиками тысяч разных эталонов. Сделать это вручную
зачастую просто невозможно. Не гарантирует успех и компьютерная идентификация,
хотя бы потому, что базы данных по свойствам эталонов содержат информацию не
обо всех известных веществах.
Особенно трудно опознать
неизвестное органическое вещество (например, выделенное из некоторого
природного объекта). В таких случаях вначале выясняют, к какой группе
органических соединений относится исследуемое вещество. Для этого регистрируют
спектр поглощения пробы в инфракрасной области. По этим спектрам можно установить,
какие химические связи и какие группировки атомов входят в состав молекулы Х.
Полезно определить молярную массу Х элементный состав пробы (например, по
атомно-эмиссионному спектру). Учитывают происхождение пробы, ее растворимость и
прочую дополнительную информацию. Совокупность полученных данных позволяет
сделать предположения о составе пробы и
составить краткий список наиболее «подозреваемых» соединений. Далее однотипные
физические характеристики пробы и каждого из “подозреваемых” сопоставляют, как
и при решении предыдущей задачи. И, наконец, делают окончательный вывод о
составе пробы.
Проверка присутствия некоторых компонентов в пробе сложного состава. Эти задачи встречается весьма
часто (например, при обнаружении опасных токсикантов в объектах окружающей
среды), но решают их иначе, чем задачи, связанные с опознанием чистых веществ.
Очевидно, что проба сложного состава и ее предполагаемый компонент в чистом
виде не могут совпасть по таким физическим свойствам, как плотность или температура
плавления, даже если компонент действительно присутствует в пробе. Надо использовать
характеристические свойства веществ, а именно – проверять, проявляет ли проба
специфические признаки присутствия
предполагаемого компонента.
Присутствие тех или иных элементов
в неорганических веществах обычно проверяют методом атомно-эмиссионного
спектрального анализа. Исходят из того, что каждый элемент имеет в своем
спектре определенный набор длин волн, на которых можно излучают свет его атомы. Наборы спектральных линий у разных элементов не совпадают. Конечно,
отдельные линии случайно совпасть могут, но весь набор линий не совпадает
никогда. Поэтому для обнаружения Х
проверяют, дает ли проба в соответствующих условиях излучение на длинах волн,
характерных для элемента Х[1]. Вывод о
наличии Х делают, если излучение пробы
на нескольких таких длинах волн достоверно превысит уровень фона.
Любые идентификационные признаки
должны быть:
·
характерными для определенного
компонента исследуемой пробы,
·
не совпадающими у разных компонентов
(селективными или специфическими),
·
не зависящими от количественного
содержания компонента в пробе,
·
устойчивыми, то есть не
изменяющимися в присутствии других компонентов пробы и при небольших колебаниях
условий измерения,
·
достаточно легко и точно
измеряемыми, даже при низком содержании компонента,
В некоторых методах качественного анализа каждый предполагаемый
компонент пробы опознают только по одному признаку (потенциал полуволны в полярографии, время удерживания
компонента в хроматографии). Гораздо надежнее те методы качественного анализа,
в которых каждый Х характеризуется множеством одновременно регистрируемых
признаков (например, положения и относительные интенсивности нескольких
спектральных линий Х). Одновременное случайное совпадение пробы и эталона по
нескольким признакам в отсутствие Х
маловероятно.
Анализ неизвестного вещества. Самая
сложная задача качественного анализа - установление
полного качественного состава пробы
неизвестного состава (возможно, сложной смеси множества веществ), когда никакой
априорной информации о ее возможном составе нет. Подобные задачи встречаются в практике довольно редко, например,
в криминалистике или в космохимии. Полный качественный анализ пробы сложного
состава представляет собой длительное и трудоемкое исследование, которое нельзя
провести по каким-то стандартным методикам.
Нетрудно установить элементный состав такой пробы, но выявить ее полный
молекулярный состав без разделения компонентов удается далеко не всегда.
Поэтому в ходе анализа неизвестного вещества обычно проводят последовательное фракционирование пробы,
вплоть до полного разделения всех ее молекулярных компонентов. Только после разделения
каждый из компонентов идентифицируют теми способами, которые были описаны в разделе «Опознание чистого вещества».
Количественный анализ с применением инструментальных методов
Любой
физический или физико-химический метод
количественного анализа предполагает
проведение следующих операций:
1. Определение градуировочной
функции (эту операцию часто называют градуировкой). Вид
градуировочной функции известен из теории соответствующего метода.
Градуировку проводят опытным путем,
измеряя в выбранных условиях сигналы подходящих образцов сравнения (ОС). Затем
строят градуировочный график на миллиметровой бумаге.
2.Измерение аналитического сигнала. Измерение
сигналов всех проб и всех образцов сравнения должно проводиться с помощью одной
и той же аппаратуры, тем же исполнителем
и в тех же условиях. Полезно
провести несколько повторных измерений сигнала
каждой пробы и в дальнейших расчетах использовать средние значения.
3.Расчет содержания Х ведут по величине аналитического сигнала,
полученного для данной пробы. Выбирая тот или иной способ расчета, учитывают
вид градуировочной функции, количество
эталонов, которые использованы для градуировки, состав этих эталонов и
требуемую точность анализа.